The Bacterial Replication Machine
(Click images and movies to expand)
Contents:
DNA Polymerase
The instructions for the living cell are encrypted in the double-strand (ds) DNA genome. For cells to divide, and form two new daughter cells, they must first duplicate all of the instructions in their DNA. DNA polymerase (Pol) is the enzyme that makes new DNA. It does so by using one strand as the template to make a new strand resulting in a dsDNA. Thus the two strands of DNA, once separated, serve as templates for DNA Pols to make two identical new copies of dsDNA molecules. The cell can then divide to form two daughter cells, each with their own complete set of DNA instructions.

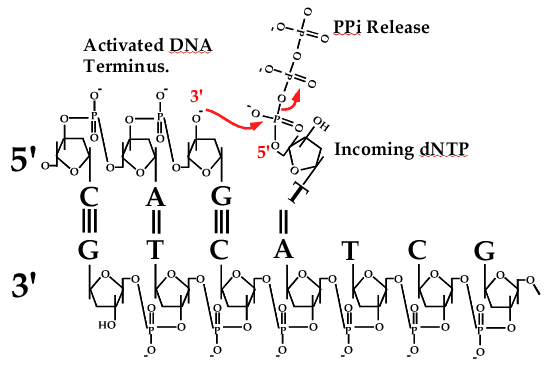
Figure 1. Chemistry of DNA polymerases. Left, picture of Arthur Kornberg. Right, chemistry of the DNA polymerase reaction. Polymerase matches the dNTP to DNA (in this case, incoming dT to template dA), then activates the 3′ OH of one DNA strand and attaches it to the 5′ triphosphate of the incoming dNTP. The reaction releases two phosphates and adds one nucleotide (dNMP) to the DNA.
The first DNA polymerase, and how it works was discovered by Arthur Kornberg, who formed the world renown Biochemistry Department at Stanford University (see photo in Fig. 1, left). E. coli Pol I binds dNTPs and matches them to the DNA template then links them together, and repeats this action over and over until it makes a long strand of DNA. The reaction is shown in Fig.1, right. Each dNTP contains a nucleotide base (either T, C, G or A) connected to a ribose sugar that contains 3 high energy phosphates at one position. The DNA polymerase activates the DNA at the 3′ OH group of the last ribose on the chain, and this causes it to attack the high energy 5′ triphosphate of a new dNTP, releasing two phosphates and extending DNA by one nucleotide. This reaction is repeated over and over again millions of times to duplicate a bacterial genome, and billions of times to make a human cell divide.
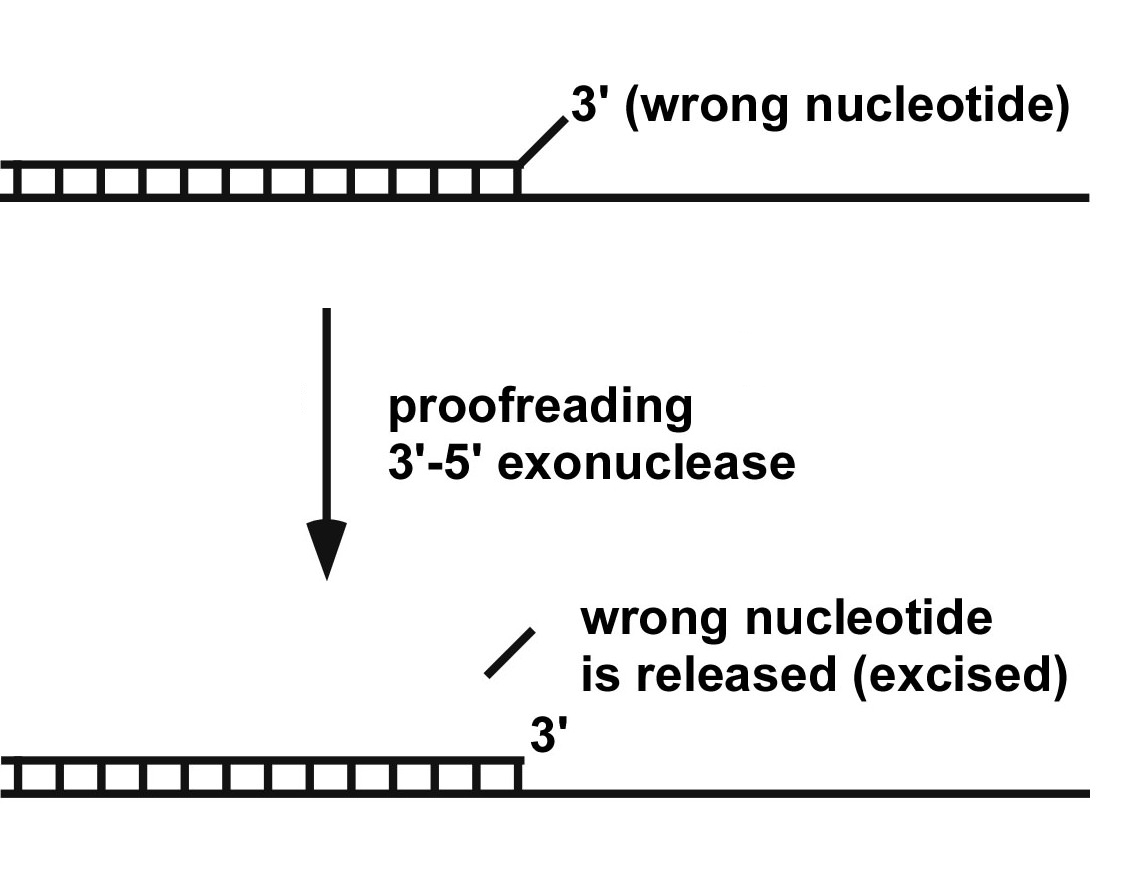
Figure 2. The proofreading exonuclease reaction. If DNA polymerase puts in a wrong incoming dNTP, the 3′ dNMP nucleotide will be mismatched. The polymerase is very slow to extend a 3′ mismatched dNMP, and the 3′-5′ exonuclease (a separate site in most DNA polymerases) finds the mismatched 3′ nucleotide and excises it from the DNA, allowing the DNA polymerase to continue.
DNA polymerases sometimes make mistakes, and if left unchecked, these become mutations. Mutations are nearly always harmful, but in rare instances they actually offer a benefit. It is this process of rare beneficial mutations that drive the process of evolution. Without mutations, we would still be single cell organisms living in the muck. Of course, mutations have their bad side too, causing cancer and all kinds of other maladies. Most of the mistakes made by a polymerase are removed before the polymerase takes another step forward. This is because DNA polymerases usually have proofreaders which are exonucleases that remove mistakenly added nucleotides so the DNA polymerase can try over again. The proofreader exonuclease is very slow compared to the polymerase. But if the polymerase puts in a wrong nucleotide base, the base is “mismatched” to the DNA template and polymerase has a very hard time to go forward. In this instance, the slow proofreader has plenty of time to remove the mismatched base (i.e. abbreviated as “dNMP”). The proofreading reaction is illustrated below. Once the mismatched wrong dNMP is removed, the polymerase continues to move forward. In this way, most of the DNA is made correctly, and only makes about 1 in a million incorrect dNMPs. These few incorrect dNMPs can result in a mutant cell, but the cell has many other DNA repair processes – so most incorrect dNMPs are corrected before a cell divides (but not all).
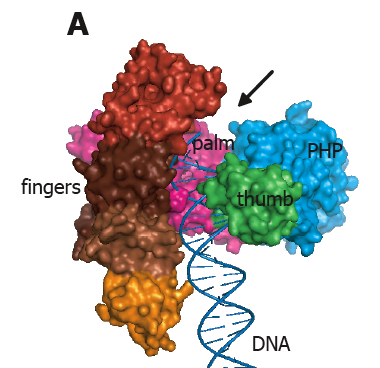
Figure 3. X-ray structure of DNA polymerase III of E. coli. The fingers, palm and thumb domains are colored and labeled. The blue domain labeled PHP is unique to Pol III; it is the proofreading nuclease in some bacterial Pol III enzymes, but not the E. coli Pol III. The PHP domain of E. coli Pol III binds the epsilon subunit, which is a 3′-5′ proofreading exonuclease. This crystal structure does not contain DNA, but the DNA is modeled into the structure (arrow). From: Lamers MH, Georgescu RE, Lee SG, O′Donnell M, Kuriyan J (2006). Crystal structure of the catalytic alpha subunit of E. coli replicative DNA polymerase III. Cell, 126, 881-92.
What do DNA polymerases look like? X-ray crystallography is the main method used to determine protein structures. The first DNA polymerase structure was of E. coli Pol I (the same enzyme Kornberg discovered), solved by Tom Steitz. Pol I has the shape of a right hand, with regions that mimic the palm, fingers and thumb of a human hand. In fact, there are many types of DNA polymerases, yet they all have been shown to have a right hand shape. A right hand shape is handy to grip DNA (no pun intended). DNA polymerases usually put in more than one dNMP at a time, by sliding along DNA as they put in one base after another. Many Pols put in 10-20 dNMPs before coming off the DNA. As we will see below, the polymerase that replicates the E. coli genome, Pol III, stays on DNA while it puts in thousands of dNMPs. A picture of Pol III is shown below.
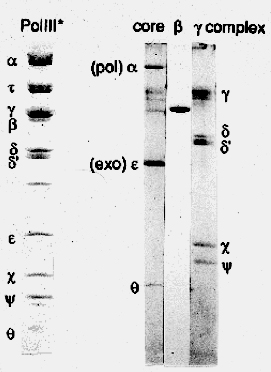
Figure 4. Subunits of E. coli DNA polymerase III holoenzyme. The first lane shows all 10 subunits that resolve by size in a SDS polyacrylamide gel. The proteins are visualized by staining with a dye (Commassie Blue) that binds to proteins. The last three lanes show subassemblies of the holoenzyme: 3-subunit Pol III core, single beta subunit, and 5-protein gamma complex. The tau subunit is not present.
Pol III is the fastest polymerase known. It contains 10 different proteins, sometimes called “accessory factors”. The largest subunit, alpha, is the DNA polymerase. The 10 subunit polymerase is referred to as Pol III holoenzyme (first lane in Fig. 4). The beta subunit can be removed to form a 9-subunit “Pol III star”. The holoenzyme can also be broken down into three components (see Fig. 4): 1) Pol III core contains polymerase/exonuclease (and theta), 2) beta subunit, and 3) the 5 protein gamma complex.
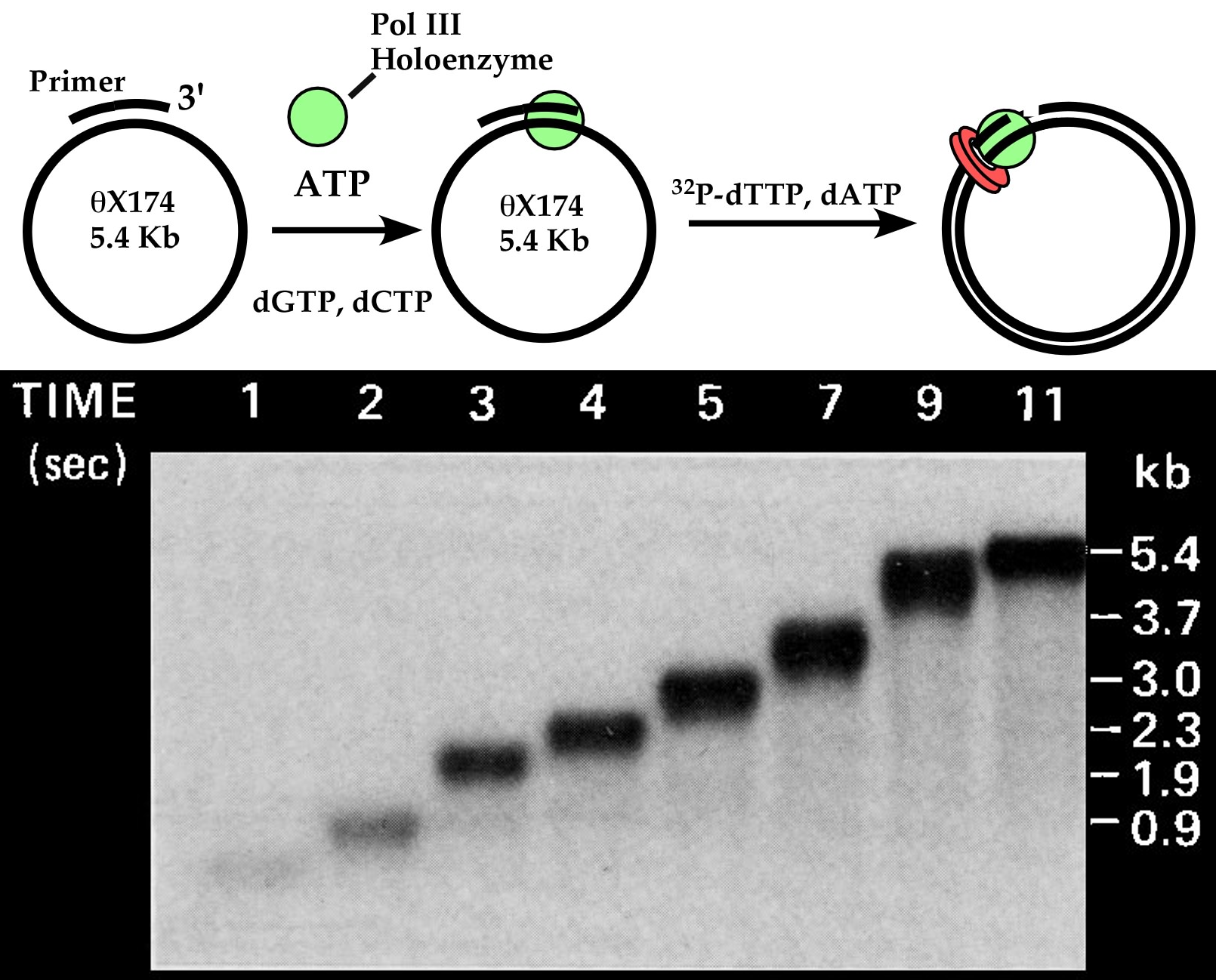
Figure 5. Speed of DNA polymerase III holoenzyme. Above, scheme of assay. The Pol III* is a complex of many proteins, which, along with beta subunit, form a tight ATP activated complex on primed DNA. Below, agarose gel of time points (in seconds) while Pol III-beta synthesizes the circular 5.4 kb ssDNA, converting it to a dsDNA circle. The experiment was performed by me, a long time ago when I was a postdoc in Arthur Kornberg′s lab. Adapted from O′Donnell ME, Kornberg A (1985). Dynamics of DNA polymerase III holoenzyme of Escherichia coli in replication of a multiprimed template. J. Biol. Chem., 260, 12875-83.
Pol III holoenzyme can use ATP to bind very tight to DNA. This tight grip to DNA results in the ability to stay bound to a DNA of many kilobases (kb) and duplicate it without coming off the DNA even once. This is referred to as highly “processive” synthesis. Pol III holoenzyme is the fastest polymerase yet discovered. An experiment demonstrating the speed of Pol III holoenzyme is shown in Figure 5. The DNA is a very large genome of a bacterial virus, the phiX174 bacteriophage genome, which is a 5.4 kb (5400 bases) circle of single-strand DNA (ssDNA)). The polymerase is first preincubated with the DNA using ATP, which gives it a tight grip to the DNA. Then the dNTPs are added, and very rapid time points are taken out of the reaction and quenched with EDTA. The EDTA removes magnesium (Mg++) by binding it very tight. Mg++ is absolutely required by DNA polymerases for catalytic activity (this is the case for most enzymes that use ATP, dNTPs, rNTPs). The “time points” range from 1 second to 11 seconds. The DNA products are then analyzed in an agarose gel, which is the method of choice to analyze the size of very large DNAs. Then the gel is exposed to an X-ray film, called an autoradiogram, to “see” the DNA products. This is because 32P-dTTP was added to the reaction to make the DNA products radioactive. As you can see from the autoradiogram, the DNA product grows longer with each time point, and is finished within 11 seconds at 30oC. For a 5400 base DNA to be complete in 11 seconds, the polymerase must add about 490 dNTPs to the primer every second!
Movie 1. DNA polymerase III acting in a highly processive fashion during synthesis. The Pol III holoenzyme is extremely processive, so much so that it does not come off DNA during the synthesis of a 5.4 kb phiX 174 ssDNA circle. Illustrated by Nina Yao, O′Donnell lab.
One reason that Pol III holoenzyme is so fast, is because it stays bound to the DNA the entire time it synthesizes the new dsDNA circle (i.e. 5400 bases). When a polymerase stays on DNA for many catalytic cycles, it is referred to as “processive”. Usually processive enzymes only stay bound to the substrate for a few, or a few tens of catalytic cycles. Staying on DNA for thousands of catalytic cycles of dNMP incorporation events is quite unusual, and would best be categorized as “highly processive”, or “extremely processive”. Click on Movie 1 to the left to see what I mean by “processive”.
How can Pol III was so fast, yet also be highly processive? It seems a contradiction, that a polymerase could achieve an extremely tight grip to DNA, yet move so very fast during synthesis. This puzzle would have to wait for a few more years, after I had my own laboratory. In fact, the finding came as a complete surprise, which is actually quite typical of most scientific discoveries. As we will see, this puzzle has a beautiful solution.
DNA Sliding Clamp
The O′Donnell lab was the first to discover a protein that completely encircles DNA. Now, many proteins are known to encircle DNA for their function. This discovery was made in two studies, first using biochemical experiments on tritium labeled beta subunit of the Pol III holoenzyme; the 3H-beta enabled its presence or absence on DNA to be followed directly. The second study used crystal structure analysis of beta, and this proved for sure that beta is shaped as a ring.
The biochemical study was a set of experiments that found that the ATP grip of the holoenzyme resided in the beta subunit, and that the beta subunit must bind DNA through the topology of the beta subunit, probably as a ring shaped protein. No protein had ever been shown to encircle DNA before, so how was this type of information obtained? The key experiments that showed that beta encircles DNA like a ring used gel filtration and radioactive beta subunit – explained below.

Figure 6. A beta dimer binds primed DNA, chaperoned by gamma complex/ATP 3H-beta was mixed with primed DNA, gamma complex and ATP, then the reaction was passed over a gel filtration column and fractions were collected. The column separates the large DNA, and 3H-beta bound to DNA (fractions 10-15) from the 3H-beta not bound to DNA (fractions 17-27). Fractions were analyzed for 3H-beta (scintillation counter), DNA (quantitated by replicating it) and gamma complex (using antibody for gamma subunit in a Western analysis). Adapted from: Stukenberg PT, Studwell-Vaughan PS, O′Donnell M (1991). Mechanism of the sliding beta-clamp of DNA polymerase III holoenzyme. J. Biol. Chem., 266, 11328-34.
The DNA used in the experiments was very large, a 5.4 kb circle of single-strand DNA (ssDNA) that was primed with a synthetic DNA oligonucleotide. Adding to its large size was the presence of single-strand DNA binding protein (SSB). SSB is a protein that binds ssDNA in a cell, and is used by all cells and all DNA polymerases at regions of ssDNA. SSB was added to the primed ssDNA circle, making it even bigger. Because of its very large size, the SSB coated primed ssDNA elutes early, in fractions 10-15, of a 5 ml column of gel filtration beads (see Fig. 6). The beads have pores, or holes, into which proteins can pass in and out of the bead. But the DNA is so large that it can′t go into the beads and therefore it goes around the beads, referred to as the “void volume” because it is the volume that is void from inside the beads. The 3H-beta is small enough to enter the pores of the beads, and therefore takes longer to come out of the column, referred to as the “included volume”. When one collects fractions of a few drops each, the void volume elutes in early fractions and the included volume elutes in later fractions. These later fractions are called the “included” fractions, because the 3H-beta goes into the beads (i.e. 3H-beta is included into them) as well as between the beads, taking more volume to travel from the top to the bottom of the column (see Fig. 6, left).
The DNA was mixed with 3H-beta (and gamma complex/ATP), and then was put over the gel filtration column. The DNA came out in the void fractions, but a dimer of 3H-beta subunits was attached to the DNA in the void fractions, while the excess of the 3H-beta came out later, in the included fractions (see Fig. 6, triangles). Antibodies to the gamma complex were used to measure the gamma complex in Western analysis of the fractions. Interestingly, the gamma complex was not present in the void fractions with DNA and 3H-beta and instead, was in the included fractions (see Western blot at the bottom of Fig. 6). This result indicates that a dimer of 3H-beta binds to primed DNA by itself, without gamma complex. Control reactions demonstrated that both gamma complex and ATP were essential for 3H-beta to bind the DNA. This result showed that gamma complex puts a beta dimer onto DNA in an ATP dependent process.
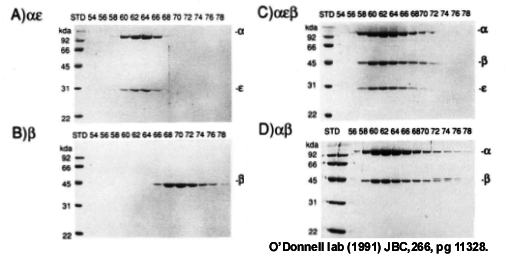
Figure 7. Beta binds directly to the Pol III polymerase. Gel filtration analysis using beads with small pores, so that the large alpha-epsilon elutes earlier than beta. Panel A, Pol III alpha-epsilon, B) beta, C) alpha-epsilon-beta, D) alpha-beta. From: Stukenberg PT, Studwell-Vaughan PS, O′Donnell M (1991). Mechanism of the sliding beta-clamp of DNA polymerase III holoenzyme. J. Biol. Chem., 266, 11328-34.
Typically, both beta and gamma complex are needed for fast and processive synthesis by Pol III core. Is the gel filtered 3H-beta-DNA complex, having no gamma complex present, sufficient to provide the Pol III core with fast and processive synthesis? The answer was yes! Adding Pol III core to the gel filtered isolated 3H-beta-DNA provided fast DNA synthesis typical of the Pol III holoenzyme. Therefore the gamma complex is only needed to get the beta subunit onto the DNA, and is not required afterward. In fact, another experiment showed that Pol III core can bind to beta directly, without the gamma complex. An experiment of this type can be demonstrated by gel filtration, using beads with very small holes, and is shown in Fig. 7 to the left. Panels A and B show the elution profiles of Pol III alpha-epsilon complex (i.e. the Pol-exo proofreader complex), and beta alone. Panel C shows that when beta is mixed with Pol III alpha-epsilon, beta binds and coelutes with Pol III alpha-epsilon, because its elution position coincides with that of Pol III alpha-epsilon. In fact, the Pol III alpha subunit alone (i.e. the polymerase) binds beta, because alpha (without epsilon) shifts the position of beta to the earlier fractions (panel D).
I also found that using gamma complex/ATP, that 3H-beta would become attached to a nicked circular dsDNA (Panel A, below). In fact over 20 beta dimers could attach to each dsDNA circle (in Fig. 3 of JBC (1991) vol 266, pg 11328), indicating that beta can “pile up” on dsDNA. About this time, I ran out of nicked circular dsDNA, but there was a nicked linear dsDNA in the freezer so this was tried instead of making more nicked circular dsDNA. But to my dismay, the 3H-beta did not bind the linear DNA. I tried it again, thinking I′d made a mistake, but still the 3H-beta did not bind the linear DNA. That night, I got to thinking that maybe beta did get on the linear DNA, but that beta was a ring shaped protein that slid off the end of DNA during gel filtration. The idea was wacky at the time, because no protein was known to encircle DNA. But I did an experiment to test this idea anyway (Fig. 8a). First, I prepared more nicked circular dsDNA. Then 3H-beta was placed onto the nicked circular DNA with gamma complex/ATP, and this time the reaction was divided in two tubes. To one tube was added a restriction enzyme that cut the DNA once and linearized it. Then both reactions were gel filtered (circle and linearized) side-by-side. The result showed that 3H-beta was bound to the circular DNA (i.e. in the void fractions), but was not bound to the linearized DNA (Fig. 8a). Yet, the 3H-beta must have been on the linearized DNA before it was cut, so the 3H-beta must have fallen off the DNA simply because of the linear shape of the DNA. This result demonstrated that the ability of beta to bind DNA depended on the topology of the DNA. One can turn this argument around and say that beta binds the DNA by the topology of the protein. Namely, that beta has the shape of a ring and can slide around and around on circular DNA without coming off, but can slide off the end of a linear DNA.
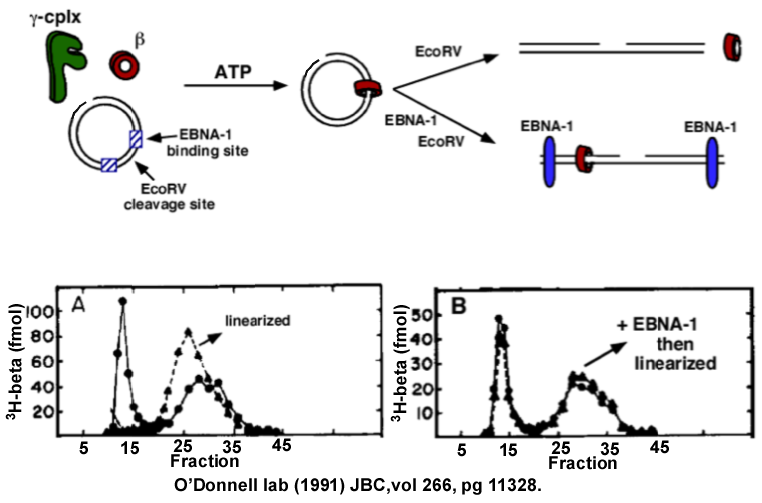
Figure 8. Beta is a circular protein. Top: scheme of the reactions. Panel A: 3H-beta is assembled onto nicked circular dsDNA, then the reaction is divided. One half is linearized with BamHI, then both reactions are loaded onto separate gel filtration columns. 3H-beta is retained on the circular DNA (void fractions 10-15), but dissociates (slides off) from the linearized DNA and all the 3H-beta elutes in the included fractions (fractions 25-35). Panel B: 3H-beta is placed on circular DNA, then EBNA1 is added, which binds two sites on the DNA. The reaction is then divided, one half is linearized between the EBNA1 sites using EcoRV. The gel filtration result shows that 3H-beta is prevented from sliding off the ends of the linearized DNA by the EBNA1 protein blocks. Adapted from: Stukenberg PT, Studwell-Vaughan PS, O′Donnell M (1991). Mechanism of the sliding beta-clamp of DNA polymerase III holoenzyme. J. Biol. Chem., 266, 11328-34.
Upon seeing this result, I knew beta was a ring that encircles DNA and slides along it. But as a further test, one would predict that a circular protein could be prevented from sliding off DNA by putting protein blocks on the DNA ends. To test this, two sites for a DNA binding protein called EBNA1 were cloned into a DNA plasmid, then EBNA1 was added and then the DNA was cut between the two EBNA1 proteins. This “protein blocking experiment” showed that 3H-beta was retained on the linear DNA by the EBNA protein blocks, as predicted if beta were a sliding ring!

Movie 2. John Kuriyan solved the crystal structure of the beta clamp. The movie of beta shows the architecture of the ring in a “ribbon representation” having alpha helices lining the inside and beta sheets lining the outside perimeter. The DNA is modeled into the center of the structure. From: Kong XP, Onrust R, O′Donnell M, Kuriyan J (1992). Three dimensional structure of the beta subunit of E. coli DNA polymerase III holoenzyme: a sliding DNA clamp. Cell, 69, 425-37.
We concluded the beta dimer is “shaped like a doughnut” and that it binds DNA topologically by encircling DNA and can slide along the duplex. We named it the “beta sliding clamp”. However, we wanted to know what this protein really looked like. Was it truly a circle? No protein was known to encircle DNA at that time. I talked with John Kuriyan, who is a world leading expert in X-ray crystallography, a method that can determine the atomic structure of a protein. Crystallography is a difficult and exacting method, but John and his postdoc Xiang P. Kong, solved the atomic structure of beta and he called me up to take a look (see Movie 2). I′ll never forget his words “Mike – you wanted a doughnut, and we give you a doughnut”. The final “picture” of beta is shown below. Never would I have expected it to look so beautiful and symmetric. Beta is a dimer, yet it had a wonderful 6-fold appearance, as the monomer was built from three domains, each with the same structure as one another. John Kuriyan was at Rockefeller University at the time, and has since moved to U. C. Berkeley. But we continued our collaboration for about 20 years regardless of his location.


Figure 9. Journal cover images featuring the beta sliding clamp. Left: “Cell” Inside article: Kong XP, Onrust R, O′Donnell M, Kuriyan J (1992). Three dimensional structure of the beta subunit of E. coli DNA polymerase III holoenzyme: a sliding DNA clamp. Cell, 69, 425-37. Right: “Nucleic Acids Research” Inside article: O′Donnell M, Onrust R, Dean FB, Chen M, Hurwitz J (1993). Homology in accessory proteins of replicative polymerases – E. coli to humans. Nucleic Acids Res., 21, 1-3.
There are numerous interesting facets to the molecular architecture of the beta sliding clamp. Too many to discuss here. The discovery of a protein that encircles DNA, coupled with its beautiful architecture, inspired the journal cover images shown to the right. The one on the left was the cover for the journal in which the structure appeared. The one on the right became the cover of that journal for a full year.
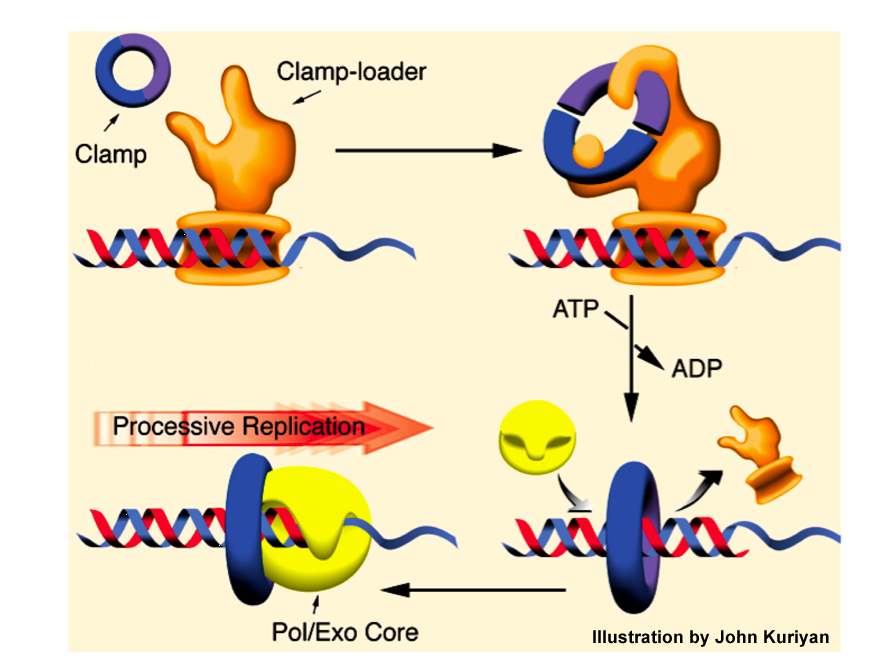
Figure 10. Illustration of how the beta sliding clamp functions. The gamma complex acts as a clamp loader, binding a primed site and using ATP to open and close the ring around DNA. The beta clamp binds to Pol III core, continually tethering it to DNA by sliding along in back of the polymerase for fast and processive synthesis. Illustration provided by John Kuriyan (prior to the crystal structures of the clamp loader and polymerase).
The diagram in Fig. 10 shows a summary of how the beta, gamma complex and Pol III core work. The beta clamp doesn′t get onto DNA by itself. It requires the gamma complex “clamp loader” which uses ATP to open and close the ring around DNA. Then Pol III core binds to the beta clamp on DNA. As Pol III synthesizes DNA (i.e. adds dNMPs), the beta clamp gets pulled along behind it, holding Pol III core to DNA and making it fast and highly processive during synthesis. The puzzle of how a polymerase binds very tight to DNA, yet moves so fast during synthesis had finally been solved.
At the time of this work, a eukaryotic system was being developed using a monkey virus called simian virus 40 (SV40). Tom Kelly′s lab had discovered how to make cell extracts that replicate the small dsDNA genome of SV40. This system enabled three labs, Tom Kelly, Bruce Stillman, and Jerard Hurwitz, to identify and purify the proteins of mammalian replication. Among the proteins were two DNA polymerase accessory factors, PCNA (proliferating cell nuclear antigen) and RFC (replication factor C). The PCNA was a trimer of a single protein (homotrimer) while RFC was 5 subunits (e.g. like the gamma complex). John Kuriyan and I guessed that PCNA would be a ring, like E. coli beta, and that RFC would be a clamp loader like E. coli gamma complex. Although there was no sequence homology between beta and PCNA, we collaborated with Jerry Hurwitz and found that subunits of RFC and the gamma complex were homologous, and therefore RFC was probably a clamp loader (O′Donnell, Onrust, Dean, Chen, Hurwitz (1993) NAR, 21, 1-3.).
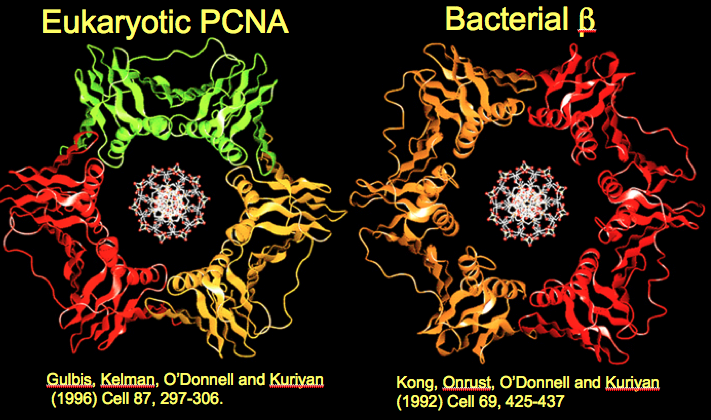
Figure 11. Human PCNA looks nearly the same as E. coli beta. Ribbon representations of PCNA and beta, with DNA modeled into their centers.
During the long collaboration of John Kuriyan′s lab with my own, nearly all the structures of the bacterial fork proteins were solved, as well as eukaryotic PCNA and RFC. Fig. 11 shows the crystal structure of human PCNA next to the E. coli beta; the two clamps are practically superimposable. The only significant difference between them is that PCNA monomers have only 2 globular domains instead of 3 domains, , and therefore it takes three PCNA monomers to form a six-domain circle. The nearly identical structure of human and bacterial sliding clamps supports the hypothesis that humans and bacteria evolved from a common ancestor cell that lived billions of years ago. The structure of the sliding clamp from this ancient cell was passed down through the eons that followed, while different multicellular animals like us evolved. We have come a long way in evolution from a single cell bacterium, but deep down inside at a molecular level, we work in much the same way.
Clamp Loader
The clamp loader is needed to assemble sliding clamps onto DNA. But how might one imagine that a clamp loader works? Why does it have 5 subunits? What does the ATP do? Does it take the circular clamp apart into monomers and reassemble them around DNA? Does it pry open just one “interface” between subunits of a clamp, and then slip DNA through the crack and into the center of the clamp? How does it bind DNA and position the DNA through the ring?
I used to think that each of the five different clamp loader subunits must have one function, and therefore there must be five functional steps to the clamp loading process. In this way of thinking, the obvious thing to do is to study each individual subunit and identify the function of each one. This way of thinking did make progress in understanding clamp loader function, but … in the end, it was the wrong way to think about the clamp loader. But first the good news –we did find something important by thinking of individual subunit functions. One obvious individual function to look for is a subunitresponsible for opening the ring up. Does one subunit do this? If so, does it take beta apart into two monomers? How would you determine this experimentally? We answered this by using radioactive beta again, but this time using 32P instead of 3H. 32P-beta can be directly visualized in agarose gels, because it exposes photographic film, making a picture called an autoradiogram. 3H does not expose film, and must be counted by a scintillation counter instead. The autoradiogram gives the scientist a visual pattern, rather than a numerical readout that must be plotted. So we assembled 32P-beta onto DNA like before, then analyzed it in an agarose gel by autoradiogram of the gel. The 32P-beta can be seen in the gel to migrate with the DNA because in the autoradiogram (first lane in Fig. 12A)m beta appears at the same position as DNA visualized in the same gel but using a ethidium bromide dye that stains DNA (first lane, Fig. 12B). The 32P-beta superimposes with the DNA in the ethidium stain, because the beta clamp is on the DNA.
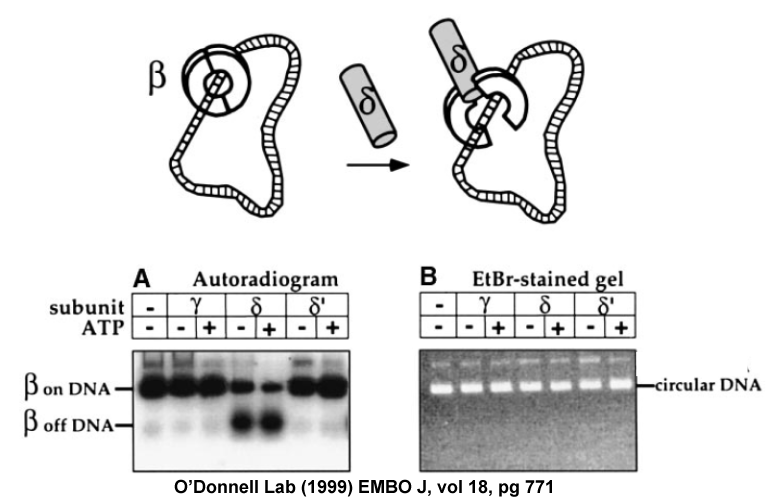
Figure 12. Experiment showing that delta subunit opens the beta ring. 32P-beta is put onto DNA by gamma complex, then gel filtered to remove gamma complex and treated with either gamma, delta or delta prime. Reactions are then analyzed in a neutral agarose gel. The gel is exposed to film to visualize 32P-beta (Panel A, left), then stained with ethidium bromide to visualize the DNA (Panel B, right). When 32P-beta comes off DNA it migrates faster than when it is bound to the large DNA. Only delta removes 32P-beta from DNA. Adapted from: Turner J, Hingorani MM, Kelman Z, O′Donnell M (1999). The internal workings of a DNA polymerase clamp-loading machine. EMBO J., 18, 771-83.
If one subunit functions to open the beta ring, it should cause the 32P-beta fall off the DNA and migrate in a different place in the gel, where beta alone migrates. We had shown earlier that only three subunits were needed for clamp loading: gamma, delta and delta prime (there are 3 gammas, so a total of 5 subunits). We examined each of these subunits for ability to take 32P-beta off DNA. The autoradiogram below showed that the delta subunit removed 32P-beta from the DNA, not gamma nor delta prime. Interestingly, only gamma binds ATP, delta and delta prime do not. Since delta does not bind ATP, yet causes 32P-beta to fall off DNA, one can reason that opening of the ring does not require the energy of ATP (since delta doesn′t use ATP). The energy source for delta to open the ring is in the binding energy of delta subunit to the beta clamp, which must cause the beta ring to open or fall apart. In our other studies we showed that delta does not take beta apart into monomers, but only opens beta at one interface, as illustrated in the cartoon at the top of Fig. 12.
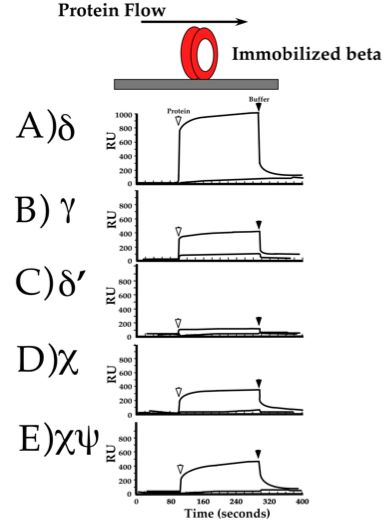
Figure 13. Surface Plasmon resonance (SPR) experiment to show protein interactions. Beta was linked to the Biacore sensor chip, and other proteins were injected into the buffer flowing over it (illustrated at the top). Each panel shows results of injecting different subunits of the clamp loader. The first arrow shows the injection of protein, and an increase in “RU” signal indicates binding to beta. The second arrow shows the end of the protein injection, and a decrease in “RU” indicates dissociation from beta. From: Leu F, O′Donnell M (2001). Interplay of clamp loader subunits in opening the beta sliding clamp of E. coli DNA polymerase III holoenzyme. J. Biol. Chem., 276, 47185-94.
For delta to unload beta from DNA, delta subunit must bind to beta. We wanted to demonstrate this binding in an experiment, and there are several ways to do this. One method is called “Surface Plasmon Resonance” (SPR) performed in an instrument called a “Biacore”. In this method a protein is first chemically linked to a surface, or “Biacore chip”. A buffer is flowed over the top of the chip it to keep it wet at all times. Then one injects into the buffer flow a second protein. If the second protein binds the protein linked to the chip, the interaction is detected as an increase in mass and this is recorded as an increase in “resonance units” (RU, which are somewhat arbitrary). When the injection of the second protein is finished, the proteins dissociate and only the protein linked to the Biacore chip remains, which is recorded as a decrease in RU. If the two proteins do not bind one another, no RU signal is observed. In the experiment below, beta was immobilized to the Biacore chip and each of the different subunits of the gamma complex was injected over the top of beta. The result shows that delta binds to beta quite well. Interestingly, some of the other subunits also bind to beta, although delta shows the strongest interaction. We will soon see why other subunits bind to beta. Although not mentioned earlier, the gamma complex has two small ancillary subunits, chi and psi, which are not required for clamp loading but are tested for beta binding in the experiment of Figure 13.
Movie 3. Structure of the delta-beta complex. Delta subunit (top) bound to a beta monomer (color, below). A second beta monomer is shown in grey and was not in the crystal structure. Adapted from: Jeruzalmi D, Yurieva O, Zhao Y, Young M, Stewart J, Hingorani M, O′Donnell M, Kuriyan, J (2001) Mechanism of processivity clamp opening by the delta subunit of the clamp loader complex of E. coli DNA Pol III. Cell, 106, 417-28.
The fact that delta opens the beta clamp begs the question of exactly how it accomplishes this. Detailed understanding of this process was achieved in our collaboration with the John Kuriyan lab. We had found that a monomeric form of beta (mutated so that it could not form a dimer) bound very tight to delta, much tighter than beta dimer binding to delta. The Kuriyan lab proceeded to determine the structure of the delta subunit bound to beta monomer. The crystal structure of the delta-beta monomer complex is shown in Movie 3. (a second beta monomer (grey) is placed in the movie).
Movie 4. The delta subunit distorts the interface of the beta clamp. The movie is an extreme close-up, showing only the interface between two beta subunits red and blue, and delta in green. Delta binds the red beta monomer and pushes the red helix, cracking the ring open. Adapted from: Jeruzalmi D, Yurieva O, Zhao Y, Young M, Stewart J, Hingorani M, O′Donnell M, Kuriyan, J (2001) Mechanism of processivity clamp opening by the delta subunit of the clamp loader complex of E. coli DNA Pol III. Cell, 106, 417-28.
The structure showed exactly how delta opens the beta ring. Delta binding to beta results in “warping” of the dimer interface, such that one interface of the beta ring can not form. This is illustrated in the “close up” in Movie 4. The structure also showed that the beta monomer “half ring” forms a shallower crescent than when it is bound to another beta monomer to form the dimer. This observation reveals that the beta ring is “strained” and has spring action stored within it, such that opening of one interface would cause the ring to open. This is discussed in more detail in the papers: (Jeruzalmi et al (2001) Cell, 106, 417-28) and (Kazmirski et al (2005) PNAS USA, 102, 13801-13806.)
Movie 5. The gamma complex pentamer. Three subunits are gamma proteins, the only subunits with ATP sites. The delta and delta prime subunits have a prominent gap between them. The place that delta binds to beta is colored yellow, and therefore beta binds underneath the clamp loader. Adapted from: Jeruzalmi D, O′Donnell M, Kuriyan J (2001). Crystal structure of the processivity clamp loader gamma complex of E. coli DNA Polymerase III. Cell, 106, 429-41.
The structure of the gamma complex clamp loader was also solved by the Kuriyan lab and is shown in Movie 5. There are three gamma subunits, and one each of delta and delta prime. The structure shows that the 5 clamp loading subunits are arranged in a ring, with a prominent gap between two of the subunits. However, the structure of the E. coli gamma complex did not reveal how it actually worked.
Movie 6. The eukaryotic RFC clamp loader bound to the PCNA clamp. The PCNA clamp is the ring below the clamp loader. The 5 subunits of the RFC clamp loader are each a different color and arranged in a ring with a gap between two subunits, like the E. coli clamp loader. Adapted from: Bowman GD, O′Donnell M, Kuriyan J (2004). Structural analysis of a eukaryotic sliding DNA clamp-clamp loader complex. Nature, 429, 724-30.
How is DNA positioned into the open clamp? How does the clamp reclose around DNA? What happens to the clamp loader after it finishes putting the clamp on DNA? These questions were addressed in a series of several papers over many years and won′t be explained in detail here. But a few of the landmarks are as follows. Firstly, our collaboration with John Kuriyan′s lab showed the structure of the eukaryotic RFC clamp loader bound to the PCNA clamp (see Movie 6). RFC has the same circular arrangement of 5 proteins as E. coli gamma complex. The PCNA clamp is positioned below RFC, as expected from study of the E. coli clamp loader.
Movie 7. DNA modeled through PCNA fits into a chamber in the center of RFC. The PCNA is in grey and the RFC is in color. The two strands of dsDNA are in pink and blue. Adapted from: Bowman GD, O′Donnell M, Kuriyan J (2004). Structural analysis of a eukaryotic sliding DNA clamp-clamp loader complex. Nature, 429, 724-30.
How does RFC bind DNA and how does it put DNA through the PCNA ring? We didn′t think we could understand this without having DNA present in a crystal structure. But in a leap of insight, John Kuriyan did something that gave a surprising revelation about these important questions. I might mention that the revelation occurred after months in which we had all spent considerable time staring at the structure, discussing it extensively and even writing a paper for sending to a journal. But at the last minute, John sat alone at the computer and did something no one had thought of. He brought a DNA molecule onto the computer screen and placed it through the PCNA ring of the RFC-PCNA structure (Movie 7). In a complete surprise, the DNA fit nice and snug right into the center of RFC! We just weren′t thinking about the center of the clamp loader as a place for DNA to be. But the fit was undeniable; the center of the clamp loader was lined with residues for DNA binding, and these residues were even conserved in the E. coli gamma complex clamp loadser. This unleashed a whole new round of biochemical and structural experiments that, over time, proved that clamp loaders bind DNA in this central chamber. The work answered the question of how clamp loaders position DNA through the ring. It was simple. The clamp loader binds right on top of the ring, and the central chamber where DNA binds is right over the central hole of the ring. Therefore, DNA within the central chamber of the clamp loader is automatically placed DNA into the middle of the ring.
Movie 8. E. coli gamma complex bound to primed DNA. Primed DNA is in green/orange. DNA binding configures the gamma complex subunits into a spiral pitch similar to the pitch of dsDNA. Adapted from: Simonetta KR, Kazmirski SL, Goedken ER, Cantor AJ, Kelch BA, McNally R, Seyedin SN, Makino DL, O′Donnell M, Kuriyan J (2009). The mechanism of ATP-dependent primer-template recognition by a clamp loader complex. Cell, 137, 659-671.
Absolute proof of DNA binding came from the structure of the E coli clamp loader bound to primed DNA (Movie 8), shown to the left. The dsDNA can be seen within the internal chamber of the gamma complex. In fact, the DNA arranged the subunits into a spiral shape having the same pitch as dsDNA. The ssDNA portion of DNA bent out the side of the gap between subunits, because the top of the clamp loaders is closed off by protein. The requirement for a sharp bend of DNA underlies the reason clamp loaders bind primed DNA ss/ds junctions, and not just dsDNA. Specifically, dsDNA is too stiff and can′t make a sharp bend, but ssDNA is very “bendable”.
Movie 9. Structure of the T4 clamp loader bound to an opened clamp and with DNA positioned through the clamp. The clamp is in green, DNA in pink, and the clamp loader is in blue and purple. Adapted from: Kelch BA, Makino DL, O′Donnell M, Kuriyan J (2011). How a DNA polymerase clamp loader opens a sliding clamp. Science, 334, 1675-80.
To obtain a complete crystal structure of a clamp loader with the DNA and the clamp, we turned to the classic T4 bacteriophage system. Many labs have worked on this classic system, starting with Bruce Alberts, then Peter von Hippel, Steve Benkovic, and the late Nancy Nossal at the NIH. We purified the proteins and made sure they were active, and the Kuriyan lab determined the structure of the ternary complex of clamp loader-clamp-DNA. Movie 9 shows the structure in which the clamp is cracked open. The clamp opens in a right-handed lockwasher shape that matches the spiral surface of the clamp loader.
Movie 10. Conformational changes associated with clamp opening. The movie morphs between two structures of the T4 clamp loader, one bound to an open clamp and one bound to a closed clamp, suggesting the type of conformation changes associated with clamp opening and closing. Adapted from: Kelch BA, Makino DL, O′Donnell M, Kuriyan J (2011). How a DNA polymerase clamp loader opens a sliding clamp. Science, 334, 1675-80.
Two structures were obtained, one with an open clamp, that had ATP in all three sites (above), and one with a closed clamp that had only 2 ATP and one ADP. These two structures suggest the conformation changes the clamp loader undertakes upon going between the open and closed clamp forms, illustrated in Movie 10.
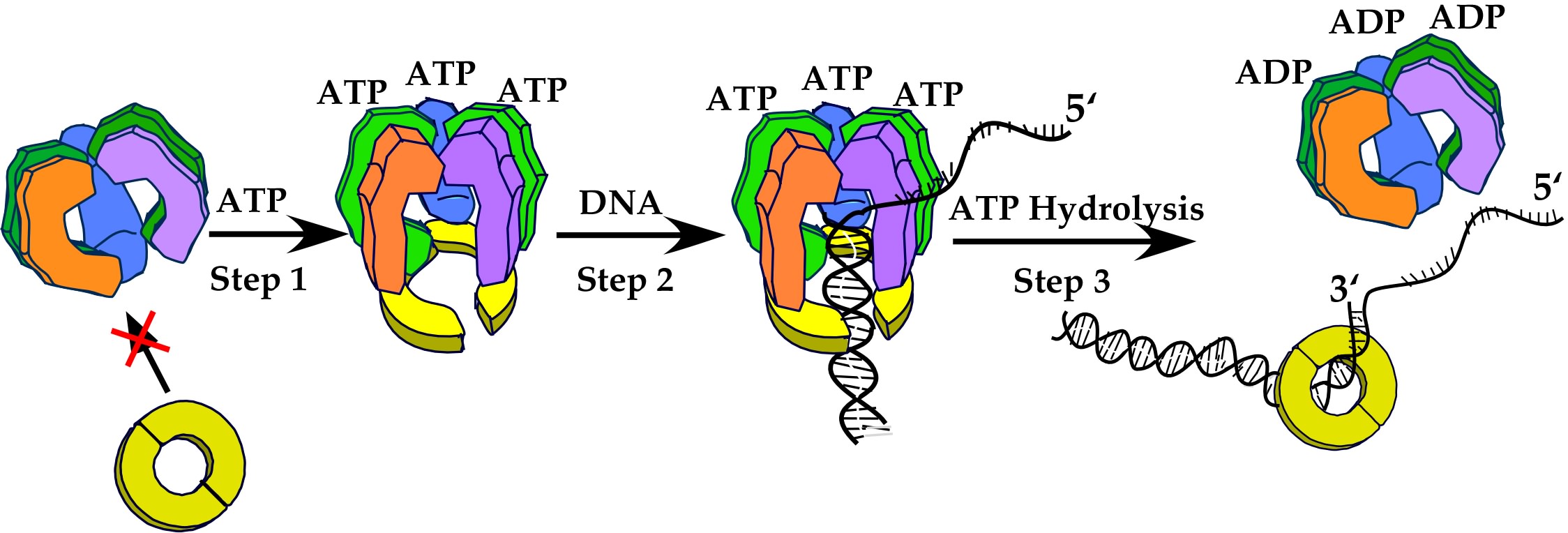
Figure 14. Overview of the clamp loader mechanism. The clamp loader is a circular pentamer that binds 3 ATP (in the gamma subunits). ATP binding changes the conformation and enables it to bind the beta clamp, opening it through interaction of the clamp with the delta “wrench” (purple subunit). Primed DNA can enter into the central chamber of the clamp loader, and this positions DNA through the opened clamp. ATP hydrolysis ejects the clamp loader, allowing the clamp to close around DNA.
In overview, after numerous biochemical and structural experiments, the clamp loader subunits don′t really work as individual pieces with separate functions, as I originally envisioned. Instead, the subunits work in unison to accomplish one specific function. Thus, all (or nearly all) the subunits bind the clamp, and hold it in an open form. All the subunits participate in binding the primed DNA. The gap between two subunits in the clamp loader is positioned right over the top of the opening bewtween subunits in the clamp, allowing DNA entry into the clamp automatically upon DNA binding the clamp loader. Once DNA occupies the central chamber of the clamp loader, the subunits are brought into a spiral conformation and the amino acid side chains forming the ATP sites are properly aligned to fire, and hydrolyze ATP. ATP hydrolysis changes the shape of the clamp loader, making it close the clamp and eject from the DNA. This leaves the closed clamp on DNA to interact with the DNA polymerase. These steps are illustrated in Fig. 14.
The eukaryotic RFC differs from bacterial gamma complex in that all 5 subunits of RFC are encoded by a distinct gene. The 5 clamp loading subuints of gamma complex are encoded by 3 genes, one for the 3 gamma, and two for delta and delta prime. However, there are two small E. coli subunits, chi and psi, that tightly associate with the clamp loader. The functions of chi and psi are distinct from the central clamp loading reaction, and we and other labs have a variety of data on what they may do.
The Replisome
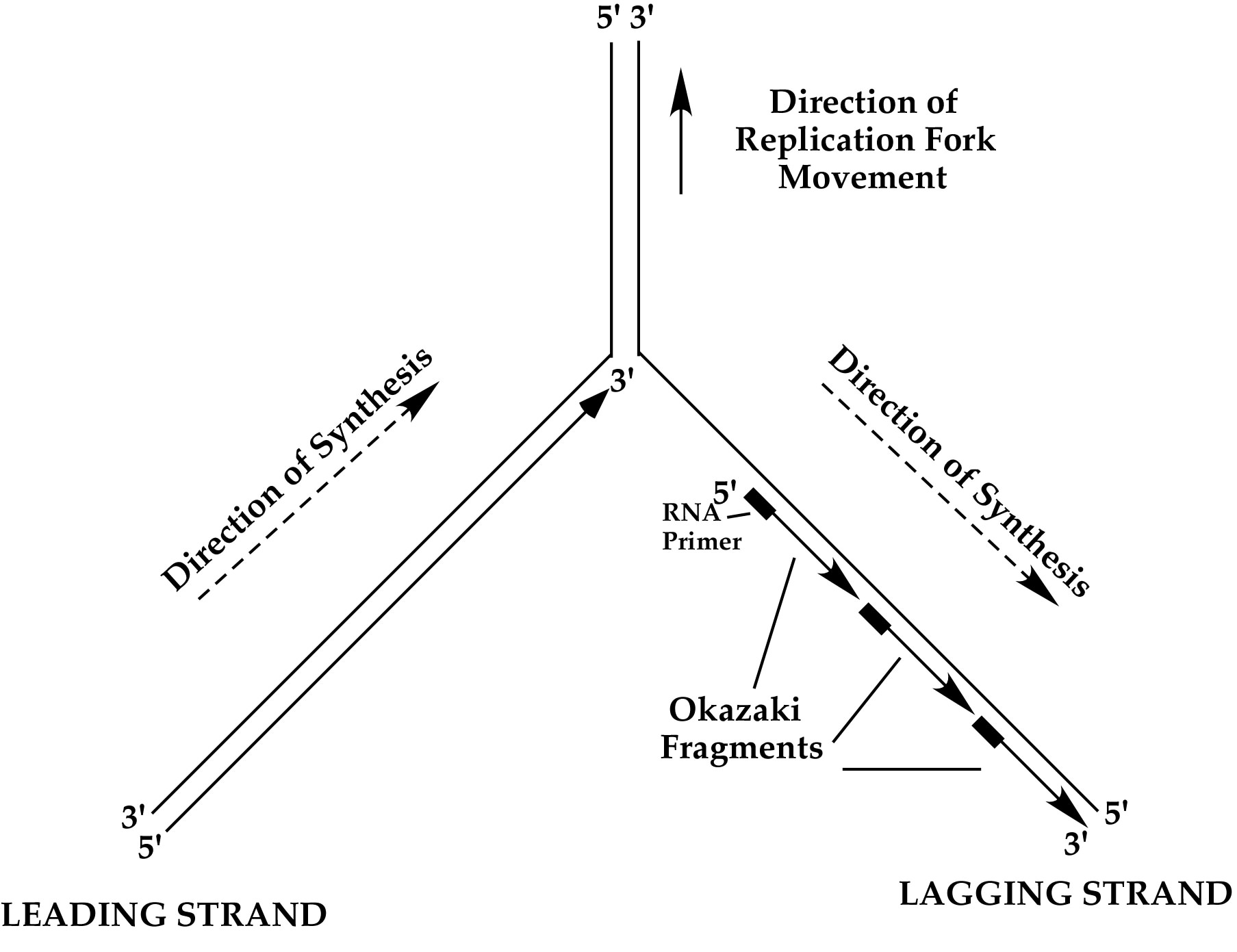
Figure 15. Replication fork DNA. The two strands of dsDNA are antiparallel, but DNA polymerases can only link nucelotide bases together in one direction. Therefore the polymerase on the “leading strand” goes in the direction of fork unwinding, and the polymerase on the “lagging strand” must go opposite the direction of fork unwinding. The lagging strand requires primase to make short RNA primers and the lagging strand is filled-in as a series of Okazaki Fragments, each extended in the opposite direction of fork movement.
To duplicate the dsDNA genome other enzymes are needed in addition to the DNA polymerase, clamp and clamp loader. One is a topoisomerase, which is needed to remove the turns in dsDNA (i.e. one turn every 10 base pairs). Another is a helicase, which uses ATP to separate the two strands of dsDNA into single-strands in order to act as templates for DNA polymerases. The point of strand separation where new DNA synthesis takes place is called a “replication fork”. The structure of a replication fork is illustrated in Fig. 15.
DNA polymerases can only extend DNA in the 5′-3′ direction (because the dNTPs are only activated on the 5′ end). Since the two strands of dsDNA are antiparallel, and DNA polymerases can only travel in the 5′-3′ direction, only one strand can be extended by DNA polymerase in the same direction as fork unwinding. This is referred to as the “leading strand”. The other strand is called the “lagging strand” and is much more difficult to replicate. DNA polymerases must repeatedly fill-in this strand in as a series of disconnected segments, referred to as Okazaki Fragments. Furthermore, DNA polymerases can not start their own DNA chains, and they require an enzyme called primase that periodically makes short RNA primers on the lagging strand during helicase unwinding. The primers are then extended by DNA polymerase into Okazaki Fragments. In E. coli, Okazaki Fragments are about 1-2 kb long. The RNA primer is later removed by an exonuclease and the gap is filled in by a DNA polymerase, then sealed by an enzyme called ligase. In E. coli the RNA primers are usually removed by a 5′-3′ exonuclease within Pol I, which simultaneously removes RNA and replaces it with DNA. But there are various ways this RNA/DNA/ligase processing step can be performed.
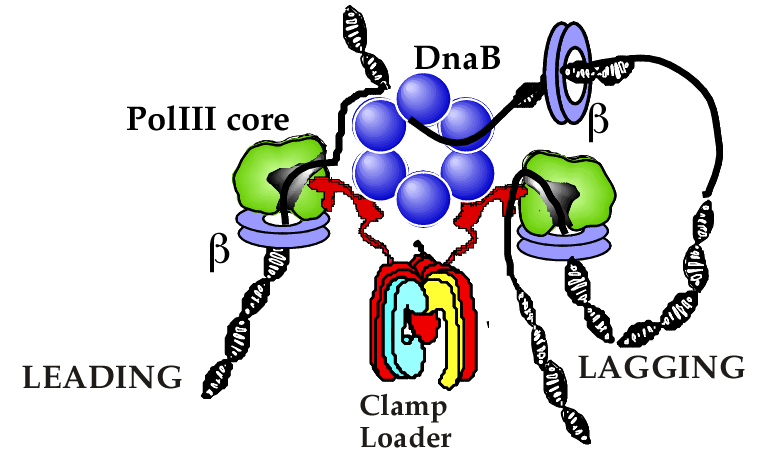
Figure 16. Cartoon of E. coli replisome organization. The clamp loader contains three tau subunits (red) that bind the DnaB helicase (blue) and Pol III core molecules (green). Each Pol III core is held to the DNA strands by a beta clamp (purple). Illustration adapted from: O′Donnell M, Jeruzalmi D, Kuriyan J (2001). Clamp loader structure predicts the architecture of DNA polymerase III holoenzyme and RFC. Curr. Biol., 11, R935-46.
Many of the proteins that separate and synthesize the leading and lagging strands at a replication fork adhere to one another to form a machine, referred to as a “replisome”. The replisome of bacteria is illustrated below. The E. coli clamp loader organizes the bacterial replisome. It contains three tau subunits that replace the three gamma subunits. The tau and gamma subunits are made by the same gene, but tau is longer than gamma. The extra length of tau gives it an additional 24kda of amino acids at the C-termini that can bind to both the helicase and Pol III core. The helicase is the DnaB homohexamer that encircles the lagging strand and uses ATP to drive unwinding of dsDNA. The tau “C-terminal arms” bind multiple Pol III cores for simultaneous replication of both leading and lagging strands. A cartoon of the replisome arrangement in E. coli is shown in Fig. 16.
Movie 11. Dynamic actions of the moving replisome. The clamp loader organizes the replisome, binding the helicase and multiple Pol III cores. As the helicase unwinds dsDNA, the leading Pol III core-beta moves continuously forward, and the primase (purple) comes in and out of the replisome to form new RNA primers. The clamp loader repeatedly places new beta clamps onto RNA primers. The lagging strand Pol III core binds the beta clamps and extends Okazaki fragments, making a DNA loop. The lagging strand Pol III core releases loops by disengaging from beta to recycle to a new beta for the next Okazaki fragment. Movie created by Nina Yao, O′Donnell lab.
The replisome is a dynamic machine, and its action is illustrated in Movie 11. The clamp loader repeatedly loads sliding clamps onto primers as they are made by primase on the lagging strand. The primase does not travel with the replisome, and this was shown by the Ken Marians group, who has also studied the replisome ands topoisomerases of E. coli. Our work on the mechanism of polymerase recycling on the lagging strand (described later) demonstrates that it hops among sliding clamps repeatedly as Movie 11 illustrates. Thus, as the lagging strand DNA Pol III core extends RNA primers into Okazaki fragments, it creates a DNA loop. Upon completing an Okazaki fragment loop, the DNA polymerase releases its clamp and lets the DNA loop go. The DNA polymerase then rebinds a new clamp on the next RNA primer to make another Okazaki fragment. This process recycles over and over during the process of replicating the DNA genome.
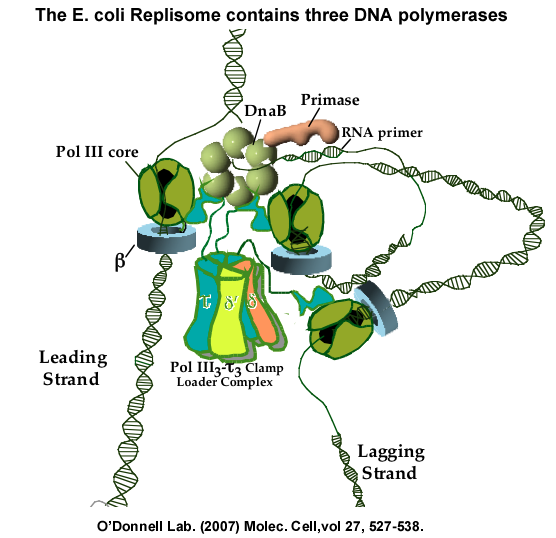
Figure 17. The triple polymerase replisome of E. Coli. Within a replisome, the clamp loader contains three tau subunits in place of the three gamma subunits. The three tau subunits bind three Pol III cores. Experiments have shown that one polymerase functions on the leading strand while two polymerases function on the lagging strand. The art work of the individual components and the DNA was conceived and drawn by Frank Leu, and later adapted to a figure from the following: McInerney P, Johnson A, Katz F, O′Donnell, M (2007). Characterization of a triple DNA polymerase replisome. Molec. Cell, 27, 527-538. And from Georgescu RE, Kurth I, O′Donnell ME (2011). Single-molecule studies reveal the function of a third polymerase in the replisome. Nat. Struct. Mol. Biol., 19, 113-6.
The E. coli replisome contains three Pol III cores, not just two. For years it was thought that the replisome would only have two DNA Pol III cores, because there are only two DNA strands to replicate. But nature does not care how we think. The clamp loader contains three tau subunits, each of which bind a Pol III core, as illustrated in Fig. 17. We first identified this by reconstituting the replicase from individual subunits, showing that it contains 3 Pol II cores (Molec Cell (2007) vol 27, p527-538). This has since been shown by other labs to be the case inside of cells. Our studies have shown that two of the polymerases work on the lagging strand (Nature Struc Mol Bio (2011), vol 19, p 113-118). On hindsight, a replisome with more than two polymerases makes perfect sense, because the lagging strand has numerous primed sites that could keep many DNA polymerases quite busy.
Key Experiments on how the Replisome Functions
Experiments that showed that Pol III core hops among sliding clamps on a lagging strand
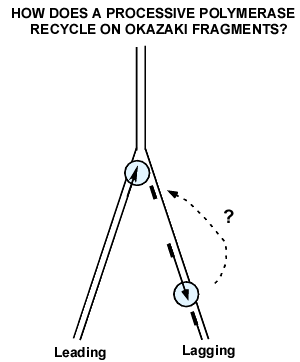
Figure 18. The polymerase recycling problem on the lagging strand. DNA polymerase III (blue) is processive because it binds very tight to DNA. But on the lagging strand, it must rapidly recycle to numerous Okazaki fragments. How does Pol III come off a completed Okazaki fragment, so that it can start a new Okazaki fragment?
Experiments that showed that Pol III core hops among sliding clamps on a lagging strand. During lagging strand synthesis, many Okazaki fragments are made, and the lagging Pol III core must be capable of rapidly transferring among the numerous RNA primed sites to generate mulitiple Okazaki Fragments (see Fig. 18). The problem is that the Pol III core is held very tight to DNA by the beta clamp, preventing Pol III core from rapid transfer from one primed site to another, as needed for multiple Okazaki Fragments on the lagging strnd. Indeed, this was a recognized problem in the field and garnered a lot of attention in how to nature overcame this problem. The simple alternative, that a different polymerase is used for each Okazaki fragment, is considered impossible given the very few Pol III molecules in the cell (10-20 molecules). Indeed, several experiments, including the single-molecule experiment shown at the end of this section, demonstrate that a single replisome is capable of extending numerous Okazaki fragments without need for additional soluble DNA polymerase.
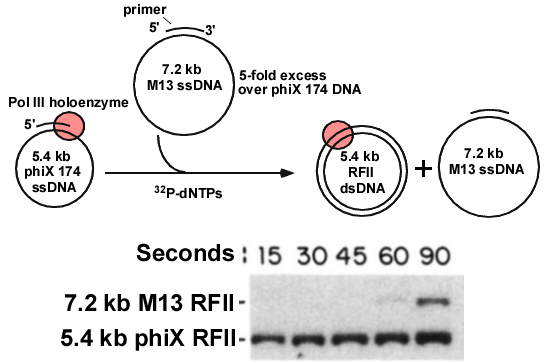
Figure 19. Pol III does not transfer to a 2nd primed DNA after finishing the1st DNA. Top: The Pol III holoenzyme was locked onto the 5.4 kb primed DNA, then a 5-fold molar excess of 7.2 kb primed DNA was added along with 32P-dNTPs. Time points were withdrawn and analyzed in an agarose gel (below). The 32P-dNTPs make the DNA radioactive. The autoradiogram of the agarose gel shows the 5.4 kb is converted to RFII dsDNA, but very little 7.2 kb ssDNA is converted to dsDNA. Adapted from: O′Donnell ME (1987). Accessory proteins bind a primed template and mediate rapid cycling of DNA polymerase III holoenzyme from Escherichia coli. J. Biol. Chem., 262, 16558-65.
The initial experiments suggested that the Pol III core-beta stayed bound to DNA that it had replicated, and would not come off. Thus, one could first bind Pol III/gamma complex/beta/ATP to lock it onto a 5.4 kb primed phiX 174 ssDNA circle, and then add a 5-fold excess of a “challenge” primed ssDNA circle of a different size (M13, 7.2 kb), and ask if the Pol III could replicate the 7.2 kb DNA after it finished making the 5.4 kb DNA. Replication was performed using 32P-dNTPs, which makes the DNA radioactive. If the polymerase finishes the 5.4 kb phiX DNA, and then transfers to the 7.2 kb M13 ssDNA, both DNAs should become radioactive, first the 5.4 kb, and then the 7.2 kb DNA. The duplex products are referred to as RFII (replicative form II). This experiment is illustrated in Fig. 19 (tip). The reactions were quenched with EDTA at various times, and then analyzed in a agarose gel to separate the smaller 5.2 kb phiX RFII dsDNA from the larger 72 kb M13 RFII dsDNA. The agarose gel was then analyzed by autoradiography (Fig. 19, bottom). The results showed that Pol III rapidly finished the 5.4 kb DNA, but was nearly incapable of transfer to, and replicating the 7.2 kb ssDNA. This suggests Pol III stayed tightly bound to the original 5.4 kb DNA after replicating it. This made sense, because Pol III has a tight grip on the 5.4 kb DNA to which ist was initially attached, so it was thought to stay bound to DNA. Presumably Pol III needs a release factor to pry it off DNA when it finishes, enabling Pol III to recycle to another primed DNA molecule.
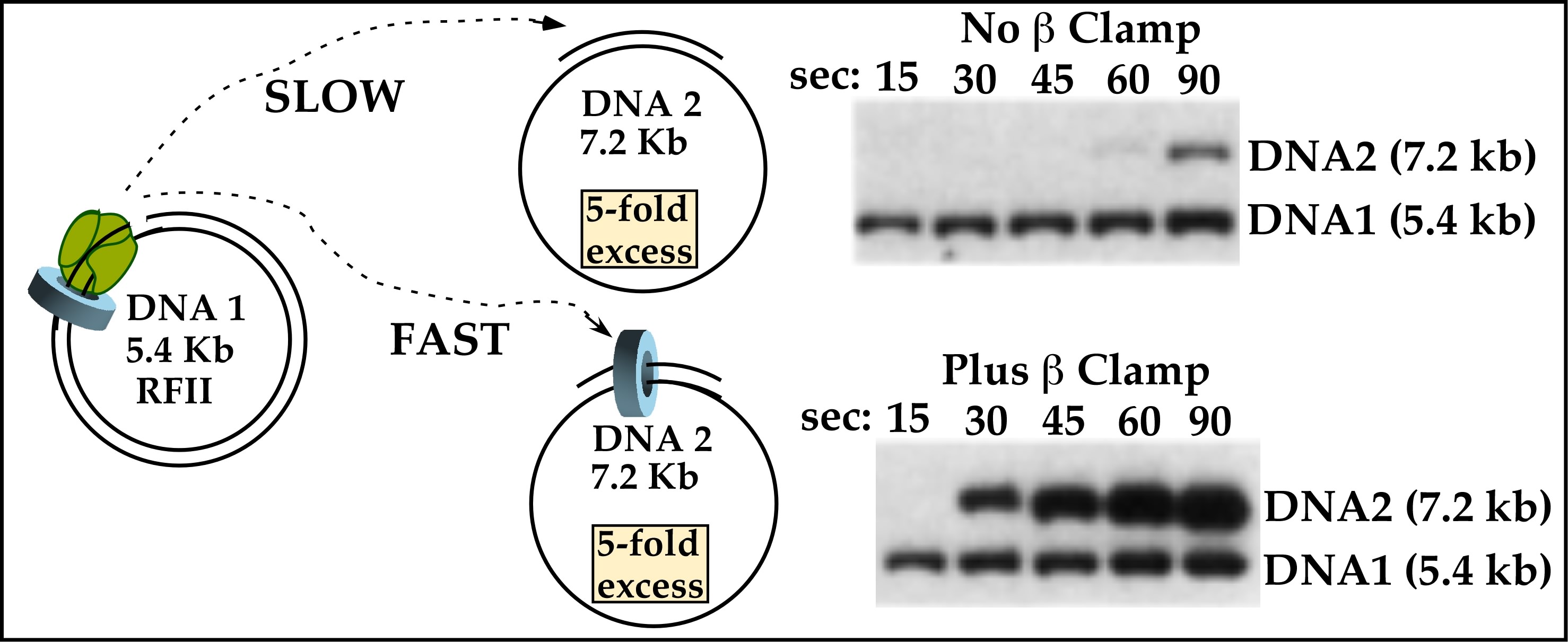
Figure 20. Pol III core rapidly hops among sliding clamps. DNA#1 (5.4 kb phiX) is treated with beta/gamma complex/Pol III core, and in a separate tube, DNA#2 (7.2 kb M13) is either untreated (top) or treated with beta/gamma complex. Then DNA#1 and DNA#2 are mixed along with 32P-dNTPs. Time points are analyzed in an agarose gel and the autoradiograms are shown. The illustration shows that after the 5.4 kb DNA#1 is replicated, the Pol III core rapidly transfers to the 7.2 kb DNA #2, but only when it has a clamp. From: O′Donnell ME (1987). Accessory proteins bind a primed template and mediate rapid cycling of DNA polymerase III holoenzyme from Escherichia coli. J. Biol. Chem., 262, 16558-65.
The first observation that revealed the process of how Pol III rapidly transfers from a finished DNA to a new DNA came from experiments just after I set up my own laboratory (see Fig. 20). I′d found that the Pol III accessory factors, beta and gamma complex, could attach to DNA without Pol III core. This gave rise to the thought that perhaps if the challenge M13 DNA were pretreated with beta/gamma complex so that the challenge DNA contained them, the Pol III core would transfer to it. To test this idea, Pol III core/beta/gamma complex was assembled on the 5.4 kb DNA circle #1 in one tube, and in a separate tube was a 5-fold excess of a challenge 7.2 kb primed ssDNA #2 that had been pretreated with beta and gamma complex (but no Pol III core). The contents of the tubes were mixed, then replication initiated using 32P-dNTPs. Time points were analyzed in an agarose gel (Fig. 20, bottom gel). The result showed 5.4 kb DNA #1 was rapidly made within 15s, and within only 30s the 7.2 kb M13 RFII dsDNA #2 was made (bottom panel, figure to the left). In fact, more and more of the excess 7.2 kb DNA #2 was made over time, indicating the Pol III core kept recycling from one replicated DNA #2 to another new DNA #2.
Movie 12. Lagging strand Pol III hops among beta clamps during lagging strand synthesis. As the fork moves forward, the lagging strand (right) exposes ssDNA. The gamma complex loads beta clamps on new RNA primers, and Pol III core transfers from the end of Okazaki fragments to new beta clamps to make multiplet Okazaki fragments. Illustrated by Nina Yao, O′Donnell Lab.
The conclusion of this experiment is that Pol III core stays bound to beta clamp and DNA during synthesis, but upon finishing DNA, the Pol III core ejects off the DNA and the beta clamp. Since Pol III core can′t replicate DNA without the beta clamp, it does not replicate the challenge 7.2 kb primed DNA#2 unless DNA#2 contains a beta clamp. In 1991 we pursued more exacting studies of the polymerase recycling mechanism, which was published in the journal Cell in 1994. The 1994 work showed that the DNA#1 is replicated to a nick before Pol III core ejects from it, and proved that beta on DNA#1 stays on the DNA after Pol III core ejects. These findings imply how the lagging strand of a replication fork is replicated. Once an Okazaki fragment is complete, the Pol III core hops off the DNA and recycles to a new beta clamp on the next RNA primer back at the fork, illustrated in Movie 12.
Experiment that shows ejection of Pol III is rapid upon finishing replication.
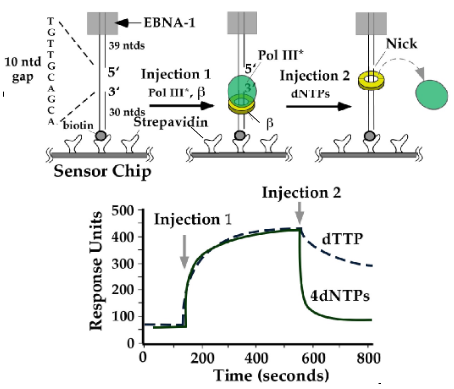
Figure 21. Biacore experiment showing Pol III comes off DNA upon finishing synthesis. DNA with a ssDNA gap is attached to a sensor chip via biotin-streptavidin. The EBNA block prevents protein exit by sliding off the top of the linear DNA. Injection 1 contains the Pol III-clamp loader (called Pol III*) and beta clamp, enabling Pol III-beta to assemble on the DNA. Injection 2 contains the dNTPs. Adding all 4 dNTPs causes Pol III to rapidly come off DNA. Adding only dTTP prevents filling the ssDNA gap, and Pol III is very slow to come off DNA. Adapted from: Leu, F.P., Georgescu, R. and O′Donnell (2003) Mechanism of the E. coli tau processivity switch during lagging-strand synthesis. Mol. Cell, 11, 315-327.
The proposal that Okazaki fragments are made by one Pol III core that hops from one clamp to another along the lagging strand makes specific predictions. One is that Pol III core should rapidly pop off DNA when an Okazaki Fragment is complete. We demonstrated this in a variety of ways. One of these methods used SPR in the Biacore machine (i.e. see clamp loader section for an explanation of SPR). A linear DNA with a ssDNA gap that contained biotin at one end was immobilized to a Biacore chip surface on which streptavidin was attached. Streptavidin binds biotin very tight, holding the DNA. The DNA also contained an EBNA1 protein bound to an EBNA site at the opposite end of the DNA, to prevent proteins from sliding off the end of DNA (see diagrams in Fig. 21). Then Pol III star-beta (i.e. Pol III holoenzyme) is assembled onto the DNA, resulting in an increase in mass on the DNA which is detected as in increase in RU resonance units (“injection 1”, below). After this, either one dNTP, or all 4 dNTPs were added. If only one dNTP was added, the gap could not be filled, and polymerase stayed bound to the beta-DNA (RU units only went down slowly). If all 4 dNTPs were added,the polymerase rapidly dissociated from DNA in about 1s (most of the RU went down in 1 s). This result suggests that Pol III comes off DNA very rapidly upon finishing replication. It is also possible the beta clamp comes off DNA, and to test this required another experimental strategy (see next experiment in Figure 22).
Experiment demonstrating that beta stays on DNA after Pol III core dissociates from completed DNA.
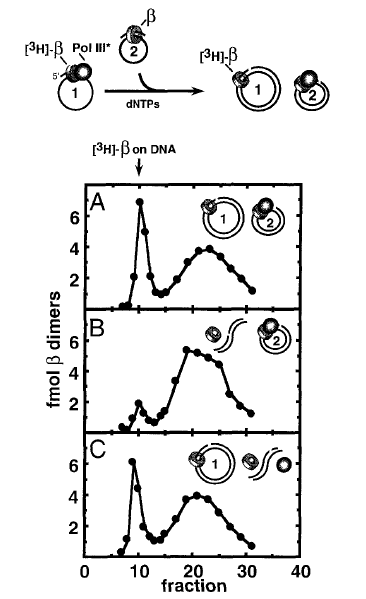
Figure 22. The beta clamp stays on DNA after Pol III leaves. Top: Scheme of the reaction. DNA#1 contains 32P-beta-Pol III complex and DNA#2 has only unlabeled beta. The two DNAs are mixed, then replicated and gel filtered either: with no treatment (Panel A), upon linearizing DNA#1 (Panel B), or upon linearizing DNA #2 (Panel C). Adapted from: Stukenberg PT, Turner J, O′Donnell M (1994). An explanation for lagging strand replication: polymerase hopping among sliding clamps. Cell, 78, 877-87.
To follow the fate of the beta clamp, (i.e. does beta stay on or come off replicated DNA), we designed an experimental stragety illustrated in Figure 23. First, 32P-beta was used to assemble 32P-beta-Pol III core onto DNA #1 (7.2 kb primed M13 DNA). In a separate test tube, unlabeled beta was assembled onto a challenge DNA #2 (5.4 kb phiX174 primed ssDNA). The contents of the two tubes were mixed and non-radioactive dNTPs were added to allow Pol III to replicate DNA #1, and transfer to DNA #2. Now we have two replicated dsDNA circles and the question is whether the 32P-beta stayed on the DNA #1, or does 32P-beta come off DNA#1 (or perhaps 32P-beta goes with Pol III to DNA#2). To answer this, we made use of the fact that beta slides off linear DNA. DNA#1 and DNA#2 have different sequences, and there are restriction enzymes specific to cut either one, without cutting the other. First (Panel A), no restriction enzyme was added, and the reaction was analyzed by gel filtration. The result showed that 32P-beta was still on DNA (elutes early in the void fractions), and therefore 32P-beta does not come off DNA when Pol III finishes replication. To determine which DNA the 32P-beta was on, the reaction was divided and treated with different restriction enzymes. The restriction enzyme used in panel B cuts only DNA #1, and the result shows the 32P-beta falls off the linearized DNA#1. Thus, beta had remained on DNA#1. Panel C used a restriction enzyme that cuts only DNA #2, and this had no effect. Overall, the results show that beta stays on replicated DNA#1, when Pol III core pops off and hops to a new beta clamp on DNA#2.
Experiment using a replisome to show that beta clamps accumulate on the lagging strand.
Movie 13. The rolling circle reaction. In this movie, the circle is spinning counter-clockwise during the reaction, while the tail with Okazaki fragments is growing and moving off to the left of the screen. The movie doesn′t show the spinning circle very well, so you need to imagine that the DNA circle is actually spinning during the movie. The DnaB helicase is purple, primase is blue, the clamp loader is green, the Pol III cores are yellow and the clamps are red. Illustrated by Nina Yao of the O′Donnell lab.
The results thus far suggest that polymerase hops among clamps on the lagging strand and since beta clamps stay on completed DNA, beta clamps are predicted to accumulate on the lagging strand as more and more Okazaki Fragments are made. A test of this prediction is to assemble the E. coli replisome on DNA (DnaB helicase, Pol III holoenzyme, primase), and ask if the beta clamps actually accumulate on the lagging strand DNA during replication. To test this, the rolling circle DNA reaction was used. For those who don′t know what a rolling circle DNA reaction is, it is illustrated in Movie 13. First, one assembles the helicase and Pol III holoenzyme (ie. contains one clamp loader, multiple Pol III cores, and beta) onto the 5′ ssDNA tail of a dsDNA circle. The DnaB hexameric helicase (purple) encircles the ssDNA tail, and the clamp (red) encircles the DNA and holds one Pol III core (yellow) onto the circle. When dNTPs and rNTPs are added, the replisome goes around and around the circle, spinning out a long DNA duplex tail. The DNA tail grows longer and longer with each time point (in the movie, the DNA exits the left side of the movie frame as it grows). Primase (blue) makes RNA primers on the growing ssDNA tail, and the clamp loader repeatedly loads beta clamps and the lagging Pol III core repeatedly extends these primers into Okazaki Fragments, leaving one beta clamp behind on each Okazaki Fragment.
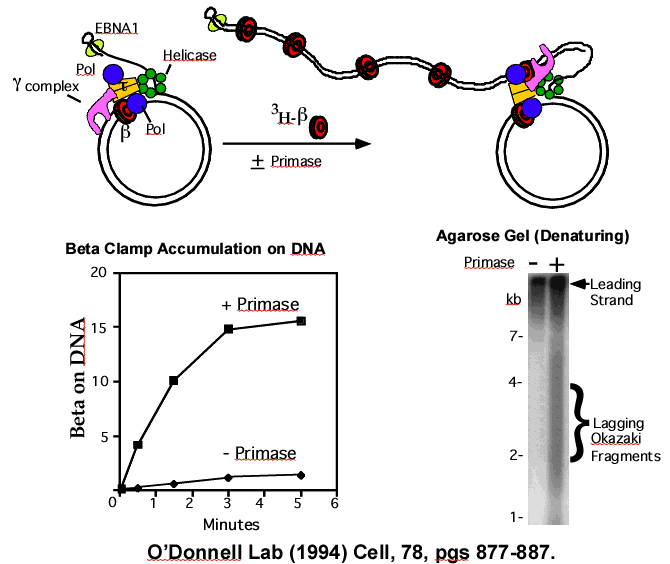
Figure 23. Rolling circle and 3H-beta clamp accumulation on Okazaki Fragments. Top – Scheme of reaction. The 5′ tail on the DNA contains EBNA1 protein to block 3H-beta clamps from sliding off the end. Replisome action results in Okazaki Fragments made on the tail. Time points are analyzed by gel filtration for 3H-beta on the DNA (squares in plot, below left). 3H-beta accumulates on DNA with time, but only if primase is present (diamonds are reactions lacking primase). The agarose gel (lower right) is of a replication reaction using 32P-dNTPs, and shows that primase is required to observe Okazaki fragments, which are smaller and migrate faster in the gel than the long leading strand. Adapted from: Stukenberg PT, Turner J, and O′Donnell M. Cell. (1994) vol 78, pg 877-887
This rolling circle system was used to test if beta clamps accumulate on the lagging strand during replication. The strategy of the experiment is shown in the cartoon at the top of Figure 23. First, the replisome was assembled on the rolling circle DNA, then gel filtered to remove any excess proteins that did not bind the DNA. The replisome-DNA complex was collected from the void fractions (i.e., left illustration). Then primase, dNTPs, rNTPs and 3H-beta were added to start the rolling circle process (right illustration). At different times, reactions were quenched with EDTA and gel filtered to see if the 3H-beta was accumulating on the DNA. The results showed that more and more 3H-beta was becoming attached to DNA during the reaction (solid squares in plot, below left in Figure 23). To test if the 3H-beta was on the lagging strand Okazaki Fragments, the reaction was performed using primase or not using primase. Without primase (plot, solid diamonds), Okazaki fragments can not be made, and the result shows no 3H-beta accumulation without primase. Only with primase was 3H-beta found to accumulate on DNA, and the agarose gel confirms that Okazaki Fragments are only made under this condition (autoradiogram, Figure 23, below right). This result supports the conclusion that beta clamps are left behind on Okazaki fragments in the wake of the replication fork. In fact, these left over clamps mark newly replicated DNA, and many labs (including our own) have now shown that these clamps bind many other types of proteins and serve a variety of functions in repair and in the process of replacing RNA primers with DNA.
Use of an alkaline gel can distanguish and visualize the leading and lagging strands. The agarose gels shown thus far are “native gels” that keep the dsDNA as a duplex. But there is another type of gel, called an alkaline agarose gel. This is the same agarose, but the buffer has 30 mM NaOH, which causes the strands of dsDNA to come apart, or “denature” into two single-strands. Once, the strands are separated, the Okazaki Fragments migrate faster than the leading strand in an alkaline gel because they are smaller than the long leading strand. The alkaline gel in Figure 23 (lower right side panel) shows the products of a rolling circle reaction using 32P-dNTPs to label the leading and lagging strand DNA. In the presence of primase, two populations of DNA are observed (second lane). The top band is the long continuous leading strand (thus it migrates slow). The smaller smear of DNA is the population of Okazaki fragments. They appear as a smear because the action of primase is somewhat random, so primers are not equidistant. This makes each Okazaki Fragment a bit different in size from one another and thus create a smear of different sizes. In the absence of primase, only the long leading strand DNA is observed, and the smear of Okazaki fragments is no longer observed (left lane).
Direct visualization of DNA as it is made by the replication machine.

Figure 24. Single-molecule microscope set-up to visualize DNA as it is replicated. Left: Microscope with CCD camera and laser fiber optics. Top right: Stage platform with valves for changing buffer flow through the flow cell. Bottom right: Flow cell, with red liquid in it. Ports for buffer input and output are separated by a narrow channel 50 microns in height. Photos: by Roxana Georgescu of the O′Donnell Lab.
We have used the rolling circle reaction to directly visualize individual DNA molecules made by the replisome in real-time. This method uses a microscope and powerful CCD camera to directly visualize the DNA as it is made and capture to it on “digital film”. To see the DNA, we used a fluorophore that gives off fluorescence when it is bound to DNA, and we used a laser to excite the fluorophore. To tether the rolling circle onto a slide that can be put under the microscope, we needed to build a flow chamber, allowing us to add proteins and nucleotides in an aqueous buffer and flow it over the immobilized rolling circle DNA. The image in Figure 24 shows the microscope, the stage platform, and a flow cell that we built with the help of the University metal shop and advice from other labs. The flow cell (lower right) contains a buffer colored red to illustrate the narrow channel where replication takes place. Buffer flows into one port, then through the narrow channel and then out the other port. The DNA is immobilized on the bottom of the narrow strip in the flow cell between the two ports.
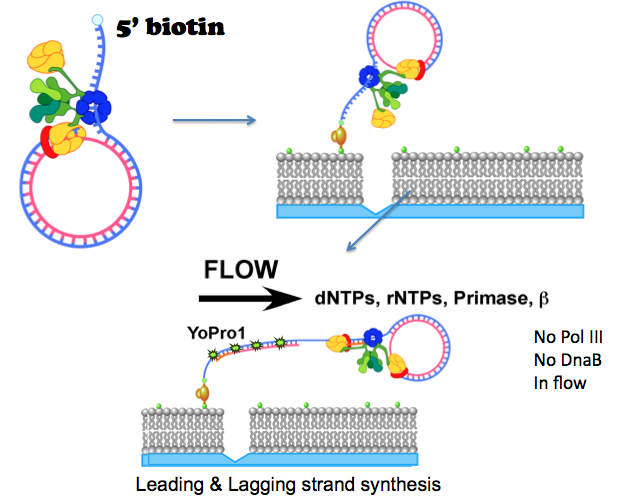
Figure 25. Rolling circle replication on a lipid bilayer in a flow cell. The DNA has a 5′ biotin and some of the lipids have a biotin (green dot). The DNA binds the lipid bilayer via a “strepavidin bridge” that binds both the DNA and lipid. The rolling circle is started by adding dNTPs, rNTPs, primase and beta in the buffer flow. The DNA tail is visualized by a fluorescent molecule (YoPro1) that binds DNA. Illustrated by Nina Yao, O′Donnell Lab.
To prevent the DNA from binding the glass of the flow cell, a lipid bilayer is formed in the flow cell by techniques we learned directly with the help of Eric Greene′s lab. The lipid bilayer allows the replicated DNA to freely extend over the lipid bilayer, but without the lipid bilayer the DNA would stick to the bare glass and form a mess. To attach the rolling circle DNA to the lipid bilayer, the DNA and some of the lipids have a biotin molecule, and they are brought together by strepavidin that holds the DNA to the lipid bilayer (see Figure 25). Lipids in a bilayer are fluid, or mobile, and so in a buffer flow, the lipid-DNA complex will migrate in the direction of a buffer flow in the flow cell. To stop the DNA from migrating completely off the slide, a small scratch is etched in the glass before making the lipid bilayer. When the DNA-lipid hits the scratch, the lipid can′t go over the scratch and DNA-lipid molecules line up on the scratch (it acts as a diffusion barrier for lipids). When dNTPs are added in the buffer flow (along with primase and extra beta), the rolling circle starts to move, generating a long dsDNA that is visualized by a fluorescent dye that binds the DNA (illustrated in Figure 25).
Movie 14. DNA molecules being made by the E. coli replisome. The field of view under the microscope shows a lipid bilayer, and a horizontal scratch on the glass surface. The lLength from top to bottom of the field of view is equal to about 300 kb of dsDNA. Buffer flow is from bottom to top, and rolling circle DNAs accumulate on the lower side of the scratch. Upon adding dNTPs (and rNTPs, primase, beta), the replisome makes DNA which is visualized by fluorescence using YoPro1 that interchelates into dsDNA. Each DNA thread contains a rolling circle DNA, located at the tip of the growing thread (i.e. at the top end). Adapted from: Yao N, Georgescu RE, Finkelstein J, O′Donnell M (2009). Single-molecule analysis reveals that the lagging strand increases replisome processivity but slows replication fork progression. Proc. Natl. Acad. Sci. USA., 106, 13236-41.
The DNA is visualized using a laser to excite the DNA-bound fluorophores, and a CCD camera takes a picture every 1 s. The movie to the left starts with a field of view of the lipid bilayer and the horizontal scratch in the cover slip. The buffer flow is from the bottom to the top, so the rolling circle DNAs line up on the bottom side of the scratch. The rolling circle DNAs can′t be seen because they are too small to bind enough fluorophore for the CCD camera to see. But upon adding dNTPs, the circles began to “roll”, and as the long dsDNA tail is made the DNA can be seen to grow longer with time. The rolling circles are located at the very tip of the growing DNA filaments. The field of view is about 300,000 bases (300 kb) long, and many DNA molecules can be seen along the width of the field of view.
There is no Pol III core, clamp loader or DnaB helicase in the buffer flow, and therefore each DNA molecule is made by a single replisome that stays on the DNA the whole time. If the DnaB helicase, or Pol III cores, or clamp loader came off the replisome-DNA, it would be carried away in the flow and synthesis of the DNA thread would stop. To determine how long these replisome components remain bound to the DNA, we can measure the DNA length when the DNA tail stops growing. Most of the DNAs are tangled and thicker than one DNA strand. But many are single, thin individual dsDNAs, and it is these DNAs from which we obtain data. Some individual dsDNA molecules are over 300 kb long, but when one plots the length of many individual molecules their average length is 86 kb. While this is much lower than the size of the E. coli genome, it is still a very long distance. It is possible that other proteins present in the cell stabilize the replisome to go much further. However, it is also possible the replisome periodically falls off DNA and constantly reassembles onto DNA to keep going. The time of replisome attachment to DNA in the cell is still an active area of research, but this experiment shows the replisome is capable of going very long distances.
Some of the Many Questions not Addressed on This Website
Why does Pol III core come off beta only when it finishes replication?
This question obviously follows from the fact that Pol III binds very tight to beta, yet Pol III core rapidly ejects from beta and DNA upon finishing a DNA. How does this happen? Although the experimental details of the answer to this question is not on this website, we have in fact looked into this, as have other laboratories in the E. coli, T4 and T7 phage systems. There are at least two routes by which this occurs. We have shown that Pol III core rapidly dissociates from beta upon completing DNA to a nick, a process called “collision release” because Pol III collides with a downstream dsDNA. Thus, there is a recognition mechanism that “tells” Pol III when it is finished and to eject from beta and DNA. There is a second process, seen only in a moving replisome, in which the Pol III core can dissociate before completing an Okazaki fragment. This is called “signal release” because the Pol III core must be signaled to eject from beta before finishing an Okazaki Fragment. The “signal” may be the torsional stress that builds during replisome movement, as shown for E. coli in (Kurth I, Georgescu RE, O′Donnell ME (2013) Nature, 496, 119-122). Work in the T4 and T7 phage systems suggests the signal may be primase or primer formation (Benkovic lab (2013) JBC 288, 20807-20816) and (van Oijen lab (2010) JBC 285, 18979-18983). The details of these mechanisms are still an area of active investigation.
How do beta clamps get recycled off DNA?
This question follows from the fact that Pol III leaves beta clamps on DNA, which accumulate on the lagging strand, one per Okazaki Fragment. The removal of beta clamps from DNA can occur in at least two processes. In one, we have shown that the E. coli cell contains an excess of the delta subunit of the clamp loader, and that delta subunit can open beta and remove it from DNA, as explained in the clamp loader section (Leu and O′Donnell (2001) JBC, 276, 47185-47189). In addition, the cell contains gamma complex that is not part of the replisome (the replisome contains tau complex, in which tau replaces gamma). The gamma complex can in fact remove beta from completed DNA, but this only works when beta is not bound by a DNA polymerase (Turner, Hingorani, Kelman and O′Donnell (1999) EMBO J 18, 771-783). This is a second route by which clamps may be jettisoned from DNA
What happens when the replisome hits an RNA polymerase?
Collisions of the replisome with a transcribing RNA polymerase can result in replisome collapse. This may seem to be a disaster, but not usually because the cell has evolved many ways to cope with this event. Given the 10-fold slower speed of transcription relative to the replisome, collisions between the two are probably frequent. When the replisome collides with an RNA polymerase on the leading strand (i.e. when transcription is in the same direction as the fork), the replisome can put a beta clamp on the mRNA/DNA junction of the transcript, and the leading polymerase can jump over and take the beta-mRNA and continue extending the leading strand (Pomerantz and O′Donnell (2008) Nature, 456, 762-766). While this may not happen in all in-line collisions, collisions of this type are favored over head-on collisions, because transcription of most genes in E. coli (and other organisms) is predominantly in the same direction as replication forks. Collisions that are head-on, when transcription is on the lagging strand, have a very different outcome (Pomerantz and O′Donnell (2010) Science 327, 590-592). In a head-on collision with RNA polymerase the replisome is blocked, but does not immediately fall apart. Instead, particular helicases can displace the RNA polymerase and let replication continue. In cases where collisions cause the replisome to fall off DNA, there are proteins that reassemble replisomes on the DNA.
What happens when a replisome hits a lesion (damaged nucleotide)?
When replisomes encounter lesions on DNA, a variety of outcomes are possible. But in almost all cases, the E. coli Pol III core stalls and cannot move past a lesion. Many labs have studied the processes that get forks past lesions. We have contributed to knowledge about one of these processes. The cell contains many different DNA polymerases, and some of these are specialized lesion-bypass polymerases that have evolved to extend DNA over nucleotide lesions of various types. We find that the replisome is a surprisingly dynamic and malleable machine. It allows translesion synthesis (TLS) polymerases to trade places with Pol III core and can utilize the beta clamp and DnaB helicase to push the replisome over the blocking lesion (Indiani, McInerney, Georgescu, Goodman, and O′Donnell (2005) Molec Cell, 19, 805-815). Pol III core can then reinsert itself to form the high fidelity replisome for continued replication.
