Eukaryotic Replication
Contents:
Introduction
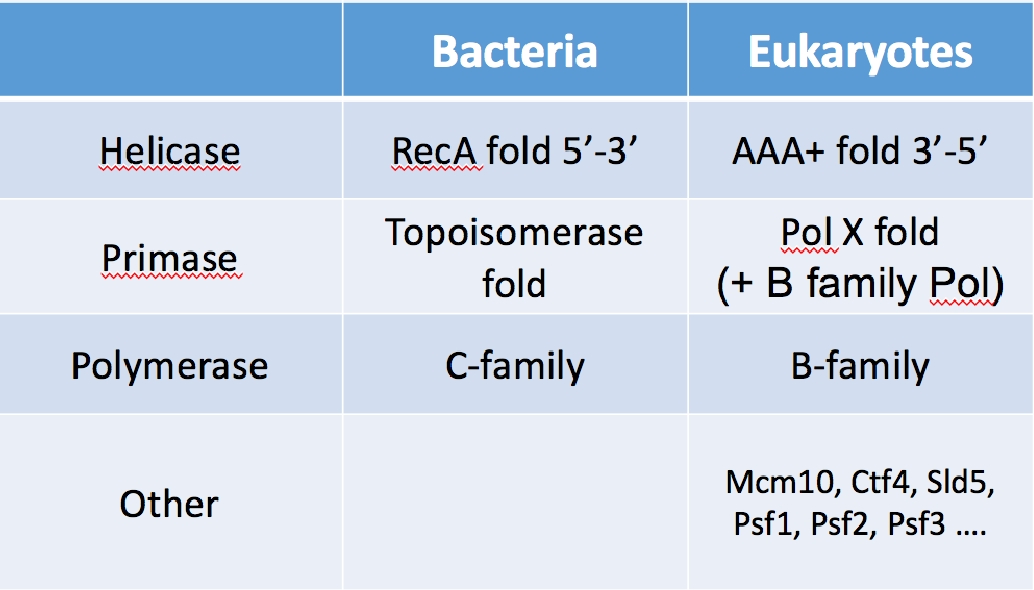
Table I. The core enzyme of the replication fork are not homologous. Thus these enzymes do not share a common ancestor in evolution. The clamps and clamp loaders are homologous, and thus were present in the last universal common ancestor.
We have recently isolated the large variety of factors that function at eukaryotic forks, and have reconstituted them into working machines. The specific function of many of the proteins are unknown other than being needed for the replication process. One may have expected to understand the eukaryotic fork from knowledge of the bacterial replisome. This is partially true, but most of the core catalytic components of the replisome are not conserved between prokaryotes and eukaryotes. The only homologous components between them are in the clamp and clamp loader, which look and operate essentially the same. But the helicase, primase, and DNA polymerases show no homology and eukaryotes contain many factors that have no bacterial homologue (see Table 1). Hence the structure and function of the eukaryotic replisome may be quite different from the prokaryotic replisome.
Biochemical Study of the Eukaryotic Replisome
There are over 30 proteins needed at a eukaryotic replication fork, and thus to study this process in detail requires they all be cloned,
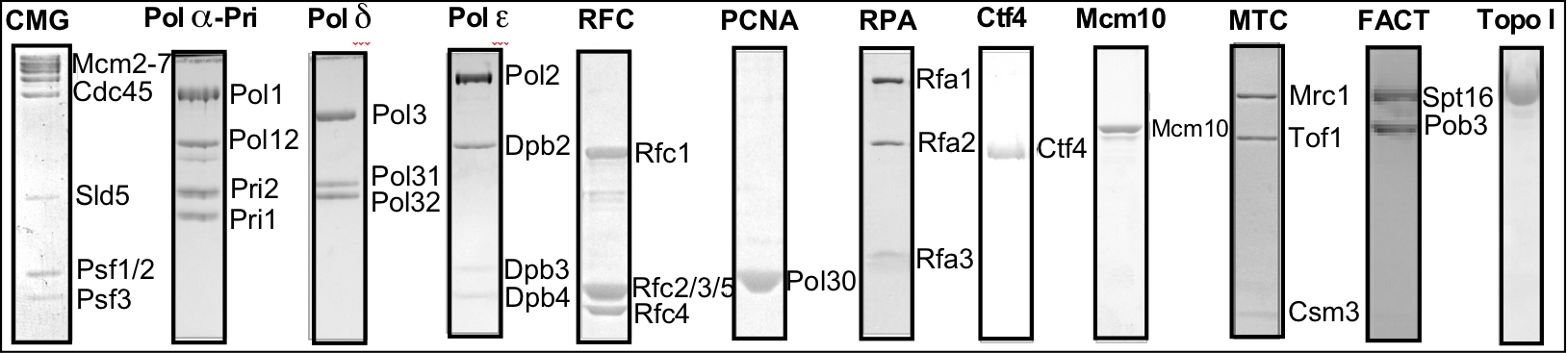
Figure 1. The proteins of the eukaryotic replisome.
expressed and purified. While difficult, my lab has worked as a team to obtain these proteins, and to reconstitute them into active complexes that form the functional leading/lagging strand replisome. The protein preparations are shown in the SDS PAGE gels in Figure 1.
The Eukyarotic Replisome
We work on the replisome of the budding yeast, Saccharomyces cerevisiae, but yeast have all the major components of the replisome in common with the human replisome. The "protein pieces" of the eukaryotic replisome are illustrated in Figure 2. The eukaryotic helicase is composed of 11 proteins called CMG, even though only 6 are illustrated in the diagram. CMG complex is an acronym coined by its founder, Mike Botchan (U.C. Berkeley, CA) that stands for Cdc45/Mcm2-7/GINS. The Mcm2-7 heterohexamer forms a ring that encircles the leading stand and tracks along it, and it may seem similar to the prokaryotic DnaB homohexamer in this way, which surrounds the lagging strand. However, the two are completely different structures and non-homologous in sequence. The Mcm subunits are based on the architecture of the AAA+ fold, while DnaB is based on a RecA folding pattern. Furthermore, to become active as a helicase, the Mcm2-7 requires five additional proteins, the Cdc45 and the 4-subunit GINS complex.
The eukaryotic primase is composed of 4 different subunits. Discovered by Bob Lehman (Stanford University, Palo Alto, CA), the Pol alpha-primase is very different from the prokaryotic primase. The two smallest subunits synthesize the RNA and form a heterodimer that defines a Pol X fold, while prokaryotic primase functions as a single subunit and its structure is based on a topoisomerase fold. In addition to the two priming subunits, the eukaryotic Pol alpha-primase contains two larger subunits, the largest of which is a DNA polymerase (polymerase alpha). The exact function of the polymerase alpha subunit is still obscure, but it extends the 7-8 nucleotide RNA primer for about 15 nucleotides, making a hybrid RNA-DNA primer. The 2nd largest of the four subunits, Pol12, is essential for life, but its function is unknown. Unlike prokaryotic primase, which is not an integral part of the replisome, the eukaryotic Pol alpha-primase appears to be integral to the replisome and is thought to travel with replication forks. One contact that secures Pol alpha-primase into the replisome is Ctf4, a homotrimer of 104 kDa subunits. Ctf4 also binds CMG, thus connecting Pol alpha-primase to the CMG helicase. Mcm10 is an essential protein that also binds Pol alpha-primase and CMG and may also travel with replication forks. Very little is known about the function of Mcm10 (despite its name, it shares no homology to the Mcm subunits of CMG).
The eukaryotic replisome uses two different DNA polymerases, Pol epsilon and Pol delta, to synthesize the leading and lagging strands. Genetic studies by the Kunkel and Burgers labs show that Pol epsilon is utilized for the bulk of the leading strand, and Pol delta for the lagging strand. Both Pols interact with the PCNA sliding clamp (see Fig. 11 in the Prokaryotic Story for a picture of PCNA). The clamp loader, RFC (Replication Factor C) is homologous to the prokaryotic clamp loader, and its clamp loading action is explained in the prokaryotic section of this website. However, RFC is not known to be an integral part of the replisome, unlike the case of E. coli, in which the clamp loader is the central organizing component of the replisome.
How do these proteins function together in a replisome?
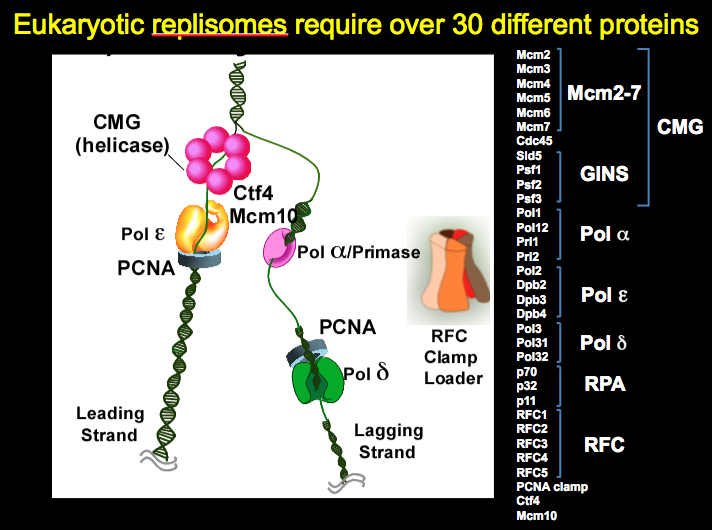 Figure 2. Eukaryotic replisomes require over 30 different proteins
Figure 2. Eukaryotic replisomes require over 30 different proteins
The eukaryotic replisome is highly regulated by modification enzymes and a variety of binding proteins. Regulation of the replisome is required for the DNA damage checkpoint response (phosphorylation, ubiquitination, and other modifications). Replisome regulation is also involved in cell cycle control, nucleosome handling and epigenetic inheritance, cohesion of sister chromosomes and other pathways involved in genome integrity. Thus, we have cloned and purified many additional proteins beyond those discussed above, and are studying how these processes interweave their actions with replisome action. Considering the rapidly evolving knowledge and complexity of these important pathways, we continue to purify new factors and modification enzymes that affect the replication apparatus in these exciting avenues of research.
What does the replisome look like?
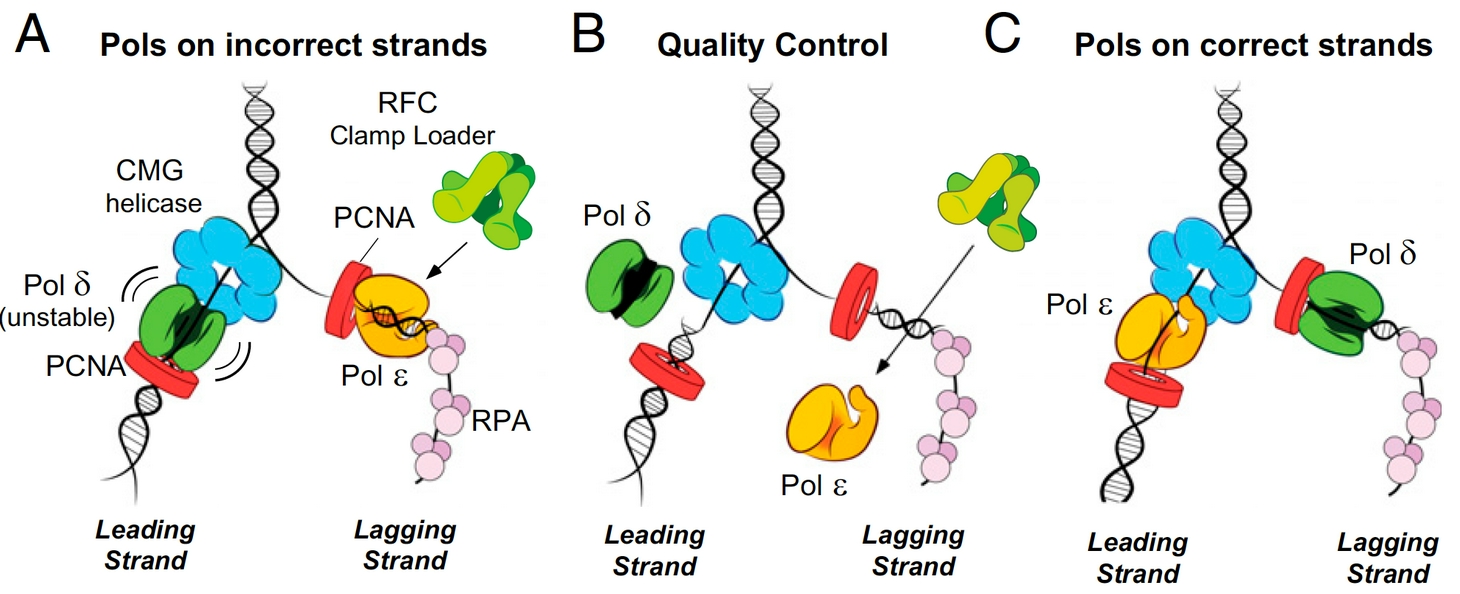
Figure 3. Quality control mechanisms that enforce Polymerase asymmetry at the eukaryotic fork. Panels a and b: If Pols epsilon or delta get on the wrong strand they are ejected by specific mechanisms. Panel c: Attractive processes that place Pols epsilon and delta on the correct strands are supported and enforced.
We have addressed several important aspects of eukaryotic replisome function by a multipronged approach, involving biochemistry, single-molecule biophysics and cryoEM structure analysis using pure proteins (e.g. see publications list). Of course, every question that is answered raises a host of new questions. But our initial studies have reconstituted a fully functional leading/lagging strand replication fork, have identified how the proteins are organized in the replisome machine, and how the DNA polymerases are targeted to the leading and lagging DNA strands. For example, we have identified a stable 15 protein complex of CMG-Pol epsilon, which we refer to as CMGE. This CMGE complex is active only on the leading strand of a replication fork DNA, supporting the evidence that Pol epsilon is the leading strand polymerase. Interestingly Pol epsilon in not able to extend lagging strand primers, which we have determined to be due to RFC inhibition of Pol epsilon, unless Pol epsilon is attached to the CMG on the leading strand. Pol delta is immune to inhibition by RFC and is fully functional on the lagging strand, but has low activity on the leading strand because it releases from DNA/PCNA upon colliding with CMG. Therefore, there are attractive and repulsive forces at work that ensure each Pol finds its correct DNA strand (summarized in Figure 3).
Architecture of the eukaryotic replisome
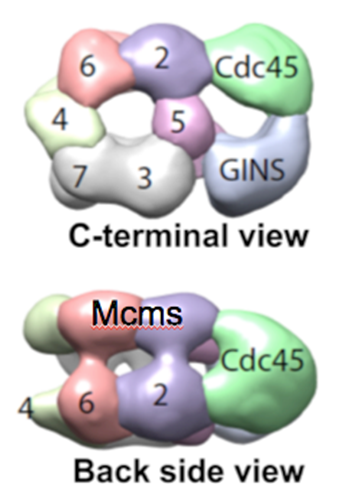
Figure 4. Structure of CMG reveals two channels. The top view shows a channel comprised of the Mcm2-7 motor proteins, and a channel made by the accessory factors and the outside surface of Mcm2 and Mcm5. The side view shows the two tiered appearance of the Mcms.
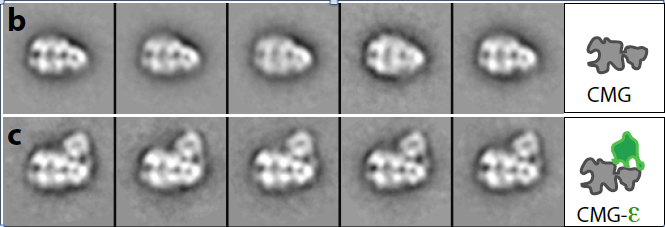
Figure 5. Structure of CMG (top) and CMGE (bottom)
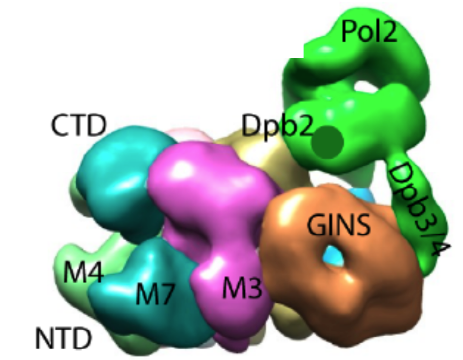
Figure 6. Pol epsilon sits on top of CMG. The green region is the density attributed to Pol epsilon. The remainder is the CMG helicase complex.
Movie 1. Rotation of CMGE (horizontal). Pol epsilon sits mainly on top of the accessory factors, Cdc45 and GINS, although it makes some contact with the Mcm ring.
Movie 2. Rotation of CMGE (vertical).The central channel of Mcm2-7 is seen. The secondary channel made by the accessory proteins is largely occluded by the Pol epsilon.
The 3D reconstruction of the replisome has been determined at low resolution by negative stain EM, in collaboration with Huilin Li (Van Andel Research Institute). We have also determined atomic models by cryo-EM. Some of these findings are explained below. The low resolution structure of CMG is shown in Figure 4.
The structures of the 11-protein CMG helicase and 15-protein CMGE (CMG-Pol epsilon) were examined by negative stain, and solved in 3D to a resolution of 15 angstroms. The structure of CMG looked essentially the same as the CMG of Drosophila, published by the Botchan/Berger/Nogales groups (see Figure 4). The yeast CMG structure, like Drosophila CMG, contains a main DNA channel through the Mcm2-7 ring.
In Figure 5, the CMGE complex can be observed, in which Pol epsilon appears as an appendage on top of CMG with multiple connections to CMG. Fig. 5 shows some 2D averages of side-view images that illustrate this point. A 3D reconstruction of the CMGE complex is shown in Figure 6.
In collaboration with Huilin Li, the cryo-EM high resolution structure of the 11-subunit CMG helicase was determined, and an atomic model derived (see Movie 3).
Movie 3. Atomic model of the 11-subunit CMG helicase. The Mcm2-7 ring (blues and greens) contains the central channel for DNA unwinding.
Architecture of the core leading/lagging replisome.
A core replisome that contained CMG helicase, Pol epsilon, Ctf4 trimer scaffold and Pol alpha-primase was built-up in stages to a final of 20 different subunits. Images of the 2D averages of the subassemblies and the final core replisome are shown in Figure 7.
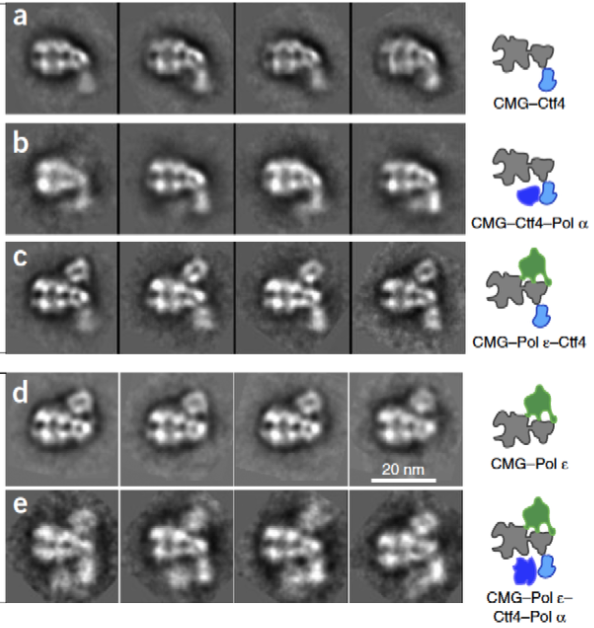
Figure 7. Buildup of the replisome. 2D averages were of the complexes indicated in the cartoons to the right. The lagging strand Pol apha-primase binds to Ctf4, under the MCM ring, and on the opposite side of the Mcms from Pol epsilon.
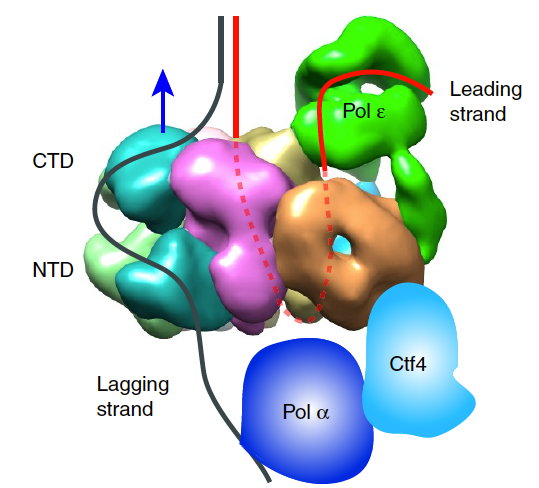
Figure 8. Architecture of the eukaryotic replisome.
Movie 4. CMG travels NTD first. The dsDNA enters the NTD face, and splits. The parental dsDNA enters the NTD face of CMG, where it splits and the unwound leading strand goes through the central channel of CMG for exit through the CTD face, where Pol epsilon binds.
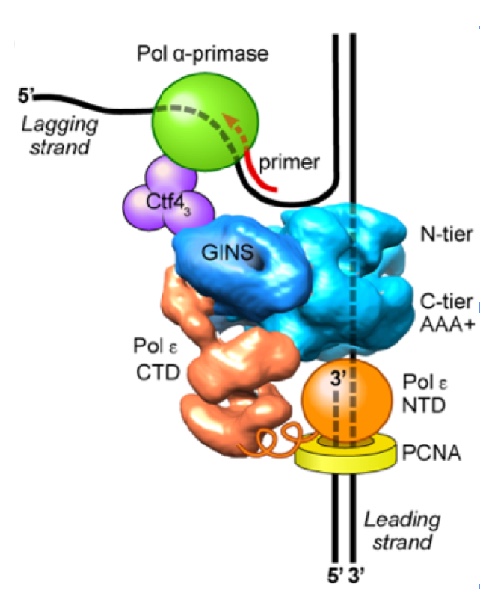
Figure 9. Architecture of the eukaryotic replisome. CMG splits the duplex DNA at the NTD face (N-tier) of CMG, enabling Pol alpha-primase (green) to prime the lagging strand as it is unwound. Pol epsilon (orange) is attached to the CTD face (C-tier) of CMG where the AAA+ motors pull the unwound leading strand through the central channel of CMG. In this geometry, Pol epsilon can continuously extend the leading strand, facilitated by the PCNA clamp (yellow).
It was long believed that DNA would enter the CTD face of CMG, because of assumptions made in the origin replication field (described later, below). Thus, in Figure 8, we show the threading of DNA through the replisome if CMG were to travel CTD-first. However, this threading would not appear to work well for a replisome, as explained later below. The replisome organization observed in Figure 8 showed that the leading Pol epsilon and lagging Pol alpha-primase were on opposite sides of the CMG helicase. CMG is known to encircle and track on the leading strand - but if parental DNA enters the CTD face of CMG - Pol epsilon, which also binds the CTD-face of CMG would be positioned above CMG helicase at the replication fork. This would require the leading strand to go through CMG, come out the NTD of CMG, and then make a 180o U-turn to reach Pol epsilon at the CTD face (i.e. see Figure 8). However, if the CMG were to track NTD-first, the Pol epsilon wound be behind CMG where it could directly extend the leading strand, and Pol alpha primase would be on top of CMG at the unwinding point for direct priming of the displaced lagging strand. Hence, we developed methods to emperically determine the tracking direction of CMG on DNA by blocking CMG during fork unwinding and enabling (by cryoEM) the first structure of a replicatiive helicase (CMG) while it was unwinding DNA at a fork. The resulting 3D cryo-EM structure of CMG at a replication fork, in collaboration with Huilin Li, showed that in fact, the CMG tracks on DNA NTD-first (see movie 4). This places the Pol epsilon at the bottom of CMG where it can extend the continuous leading strand as it is unwound, and places Pol alpha-primase at the top (NTD face) of CMG where duplex DNA is split into two single strands. This is a strategic location for Pol alpha-primase to form primers on the lagging strand (Figure 9).
Ancillary factors increase the rate of the replisome
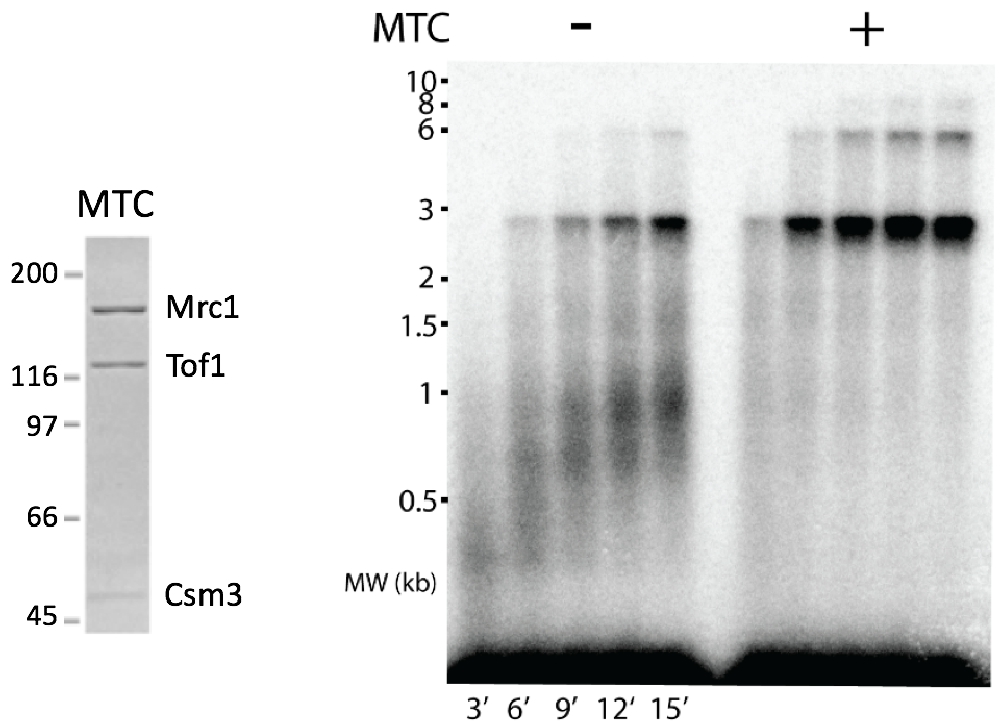
Figure 10. Mrc1 enhances replisome rate to in vivo speeds. Mrc1 is the largest subunit of the MTC (Mrc1-Tof1-Csm3) complex (see SDS PAGE gel to the left). The agarose gel to the right shows the rate of the replisome on a 3kb forked DNA + or – MTC. Addition of TC complex does not have an effect on replisome rate (not shown here).
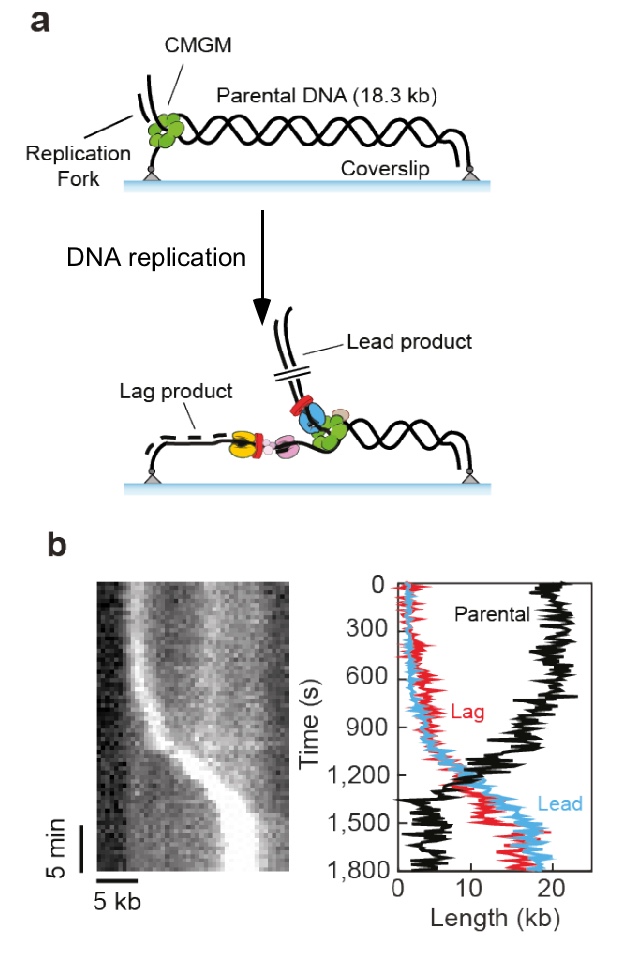
Figure 11. Single molecule set-up.a) An 18.3 kb linear fork DNA is attached at both ends. The replisome is assembled at the fork, and the leading strand is displaced during replication. b) Sytox staining of DNA shows the leading strand as a bright spot of coiled DNA traveling the length of DNA with time, shown in the kymograph (left).
The core replisome, containing CMG, Pol alpha-primase, Pol epsilon and Pol delta, RFC and the PCNA clamp only travels about 8 nucleotides/s (ntds/s). But the in vivo rate of replication is 20-25 ntds/s. Thus, ancillary factors were added to see if either Ctf4, Mcm10, Mrc1, Tof1, Csm3 could increase replisome rate. Previous cellular studies showed that Mrc1 deficient cells have slow moving forks. Thus, it was no surprise that addition of the Mrc1-Tof1-Csm3 complex increased the rate of the replisome to in vivo rates (Figure 10). The Tof1-Csm3 complex did not enhance the rate, and thus Mrc1 is the rate enhancing factor.
Dynamics of the replisome holoenzyme during replication
Single-molecule studies in collaboraton with Antoine Van Oijen (U. Woolongong, AU) showed that the replisome, formed using all the fork proteins, can replicate over 5 kb of a DNA tethered at both ends of a coverslip and in a solution containing no extra proteins (Figure 11). This work is currently in BioRxiv and under review for publication in a journal. Below is some of the data from the BioRxiv submission.
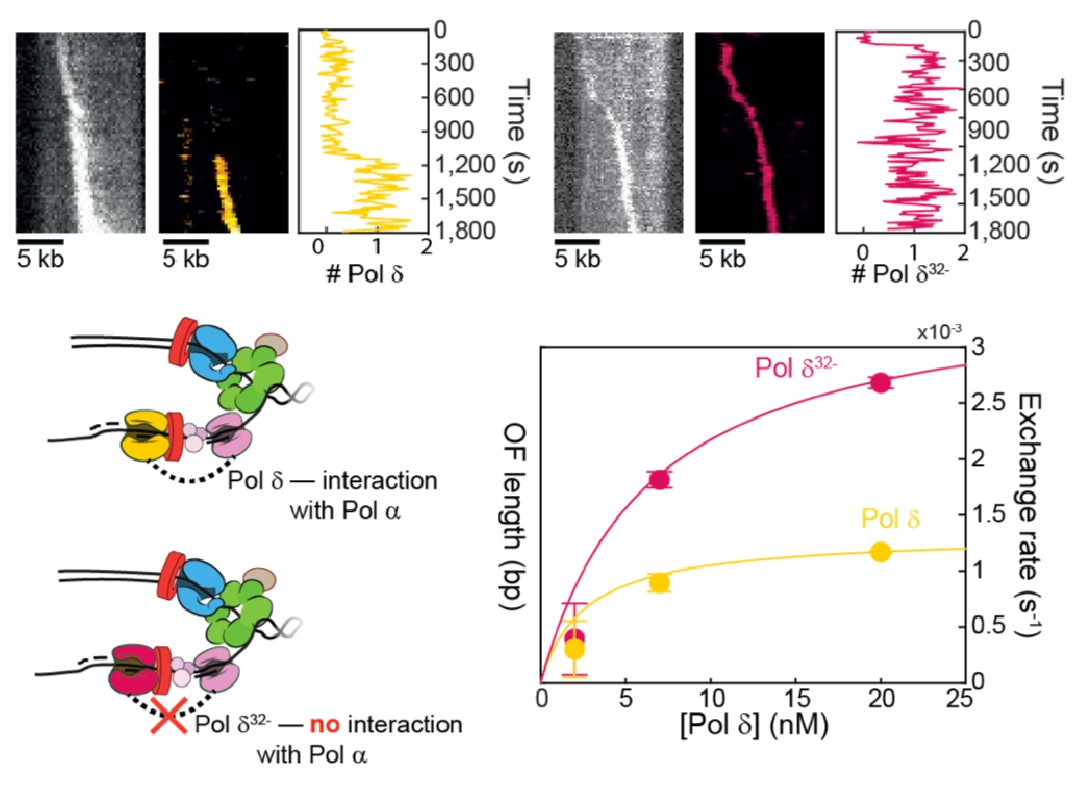
Figure 12. Pols delta and epsilon travel with the replisome.Top: Pols delta (left) and Pol epsilon (right) can travel in a processive fashion for over 5 kb with CMG. Bottom: The connection of Pol delta to the replisome is mediated, in part, by the Pol32 subunit of Pol delta.
Use of fluorescent Pols epsilon and delta showed they stay with the replisome. While this high processivity was expected for leading stand Pol epsilon, which connects to CMG and is also tethered to DNA by a PCNA clamp, the lagging strand is made as a series of short Okazaki fragments (only 150-200 bp long in eukaryotes). Hence, we were surprized that Pol delta remained attached to the replisome well over 5 kb, implying lagging strand loops are made, as in prokaryotic studies. Pol delta is reported to interact with Pol alpha primase through its Pol 32 subunit. Comparison of fluorescent wt Pol delta (yellow) and fluorescent Pol delta missing Pol32 (red), shows less stability than wt Pol delta (Figure 12).
We have examined the dynamics of the DNA Pols within the moving replisome by photobleaching in the presence of an excess of DNA polymerase in the reaction. The results show that the replisome is quite plastic, and that both Pols epsilon and delta exchange with similar kinetics and in fashion that depends on the concentration of the soluble DNA polymerase.
Origin Initiation and Implications to DNA repair
Our cryoEM structure of CMG bound to the replication fork DNA showed that CMG tracks on DNA in the NTD-first orientation. This orientation is opposite the direction long believed for the two CMGs formed at origins. Specfically, our research that flipped the orientation of CMG tracking on DNA, reveals that the two CMGs at an origin, which face N-to-N, track toward one another, instead of directed away from one another (Figure 12). This has profound implications about the last stages of origin initiation.
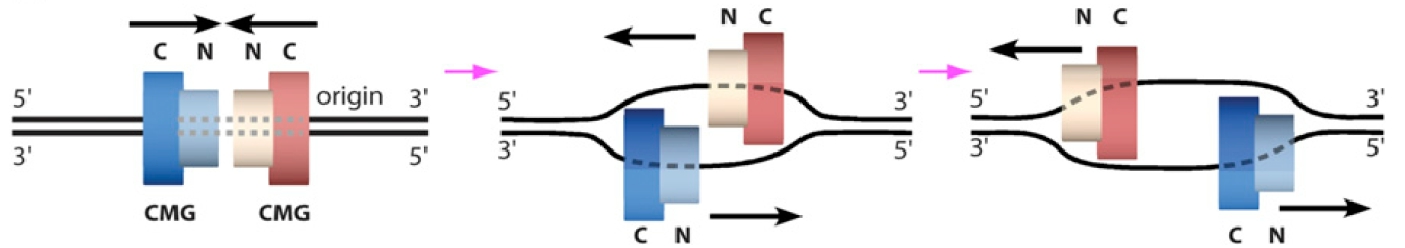
Figure 13. Two CMGs at an origin.CMGs at an origin are headed inward, NTD first, and must transition to encircle opposite strands of ssDNA in order to pass and form bidirectional forks.
Initial unwinding of dsDNA at the origin.
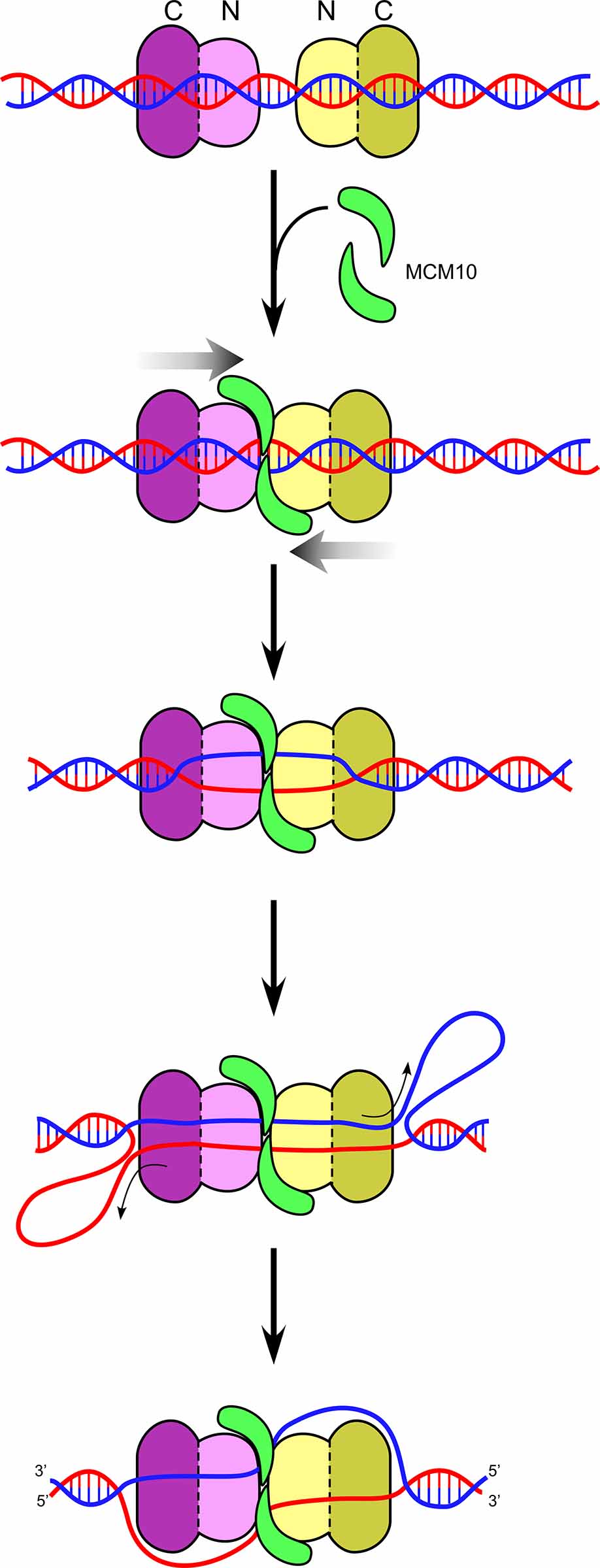
Figure 14. Head-to-head CMGs encircling dsDNA. Head-to-head CMGs are found to melt DNA in the presence of Mcm10 (green). The motors are in the CTD-tier (dark purple), and thus the opposite strands of dsDNA between the two motors is pulled upon, shearing the DNA apart. Upon unwinding sufficient DNA, shown as ssDNA loops in back of the motor domains, the CMGs need to transition from dsDNA to opposite tracking strands of ssDNA in order to pass and leave the origin.
We used specially designed DNA structures to show that CMG tracks on dsDNA, and does so with force sufficient to melt over 60bp when supplied with Mcm10. Furthermore, the CMGs track mainly on the 3’-5’ strand while encircling dsDNA (i.e. the same strand that they normally track on while acting as a helicase). When two CMGs encircle dsDNA in a head-to-head configuration (NTD-to-NTD) they use ATP to push against one another, putting strain on the duplex DNA and they cause the duplex DNA to melt up to 150 bp in the presence of Mcm10 (see Figure 14). Cellular studies show that Mcm10 is needed for these last steps of origin melting and dsDNA to ssDNA transition, and thus these studies are consistent with in vivo results. Unwinding of 150 bp of dsDNA is a sufficent length for two CMGs, each having a central channel of 110 angstroms, to eject one of the strands of unwound DNA, placing them on opposite strands of ssDNA for bone fide helicase action and enabling them to pass one another.
Replisome action during fork-reversal repair
Movie 5. Postions of Mcm10 occupancy on CMG.Mcm10 interaction cross-linking sites with CMG are shown in purple. The subunits of CMG involved are shown in colors. The NTD face of CMG is at the top in the intial frame of the movie. The DNA binding domain of Mcm10 is located near the central channel of CMG at the NTD face.
Mcm10 is known to bind the N-region of Mcm2, but beyond this contact little is known how Mcm10 binds CMG. Thus, we more precisely examined the Mcm10-CMG contact surface by collaborating with Brian Chait (Rockefeller University) using extensive cross-linking between Mcm10 and CMG, combined with mass spectrometry to identify the cross-link amino acid positions. Interestingly, the Mcm10 formed extensive cross-linking to 6 of the 11 CMG subunits, indicating an extensive interface and explaining its very tight association with CMG. The surface of Mcm10 binding to CMG by this technique is represented in Movie 5. The largest number of cross-links are to the NTD-tier of CMG, and thus would fit between the two CMGs that are oriented in a head-to-head fashion.
Transition of CMG from dsDNA to ssDNA
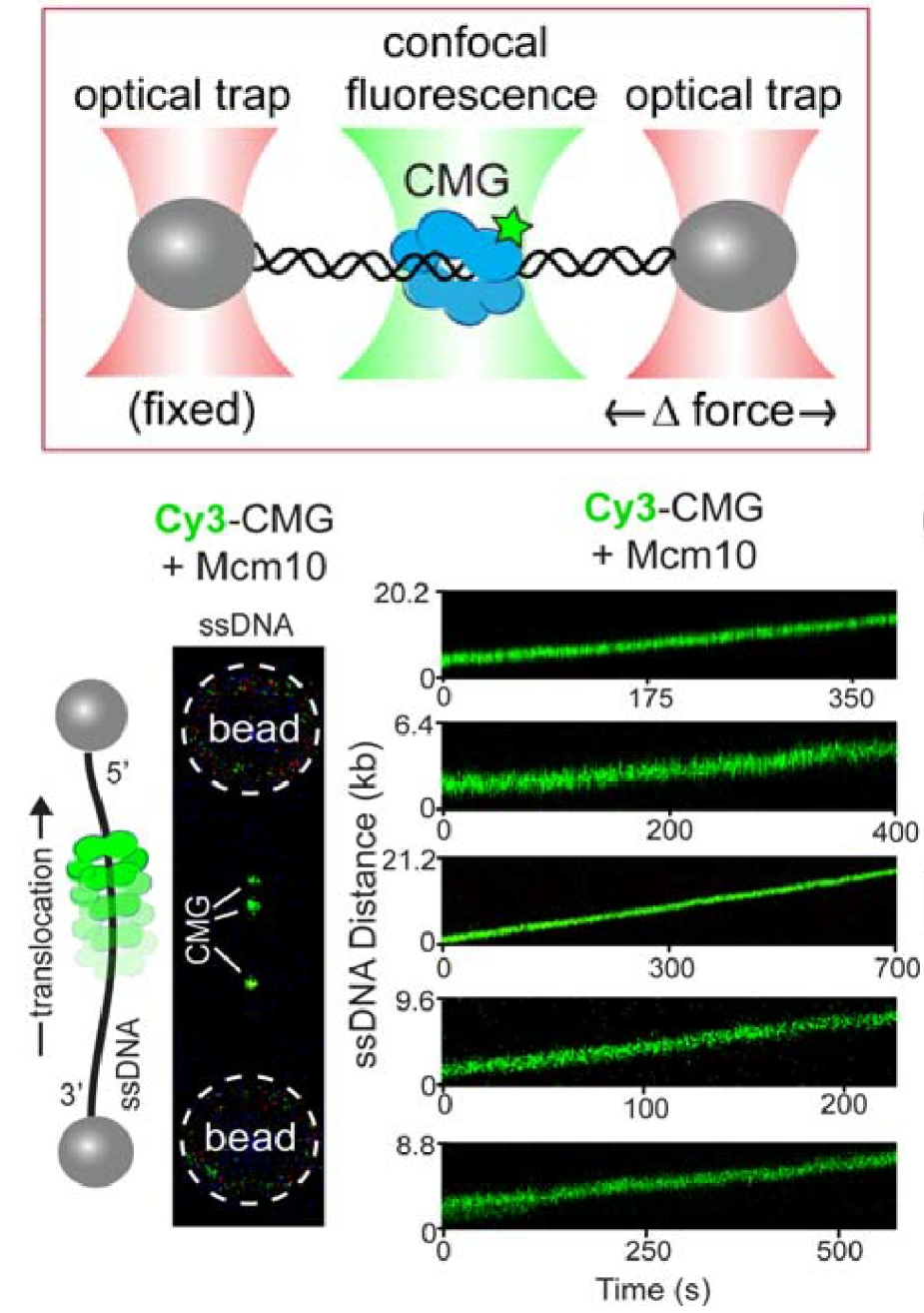
Figure 15. Single-molecule assay of a ssDNA gate in CMG.Top: Lambda phage DNA is held between two optical traps by beads attached to the ends of the DNA. Bottom: One strand of lambda DNA is help between two beads, and the “green” CMG+Mcm10 gets onto it (left), implying it has a ssDNA gate. The CMGs move along the ssDNA upon adding ATP (bottom, right).
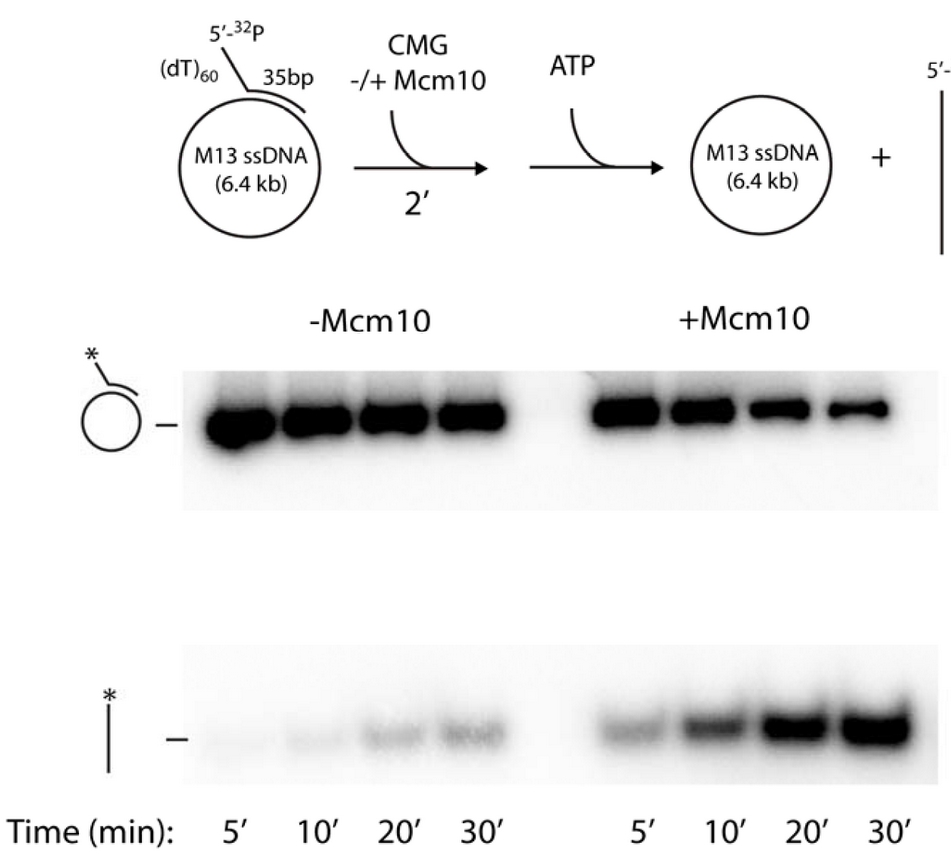
Figure 16. The circular DNA helicase assay.A 5’ 32P-primed tailed oligonucleotide is annealed to circular M13 ssDNA (top). Time course analysis in a native agarose gel shows CMG helicase can remove the 32P-oligo (bottom left). However, Mcm10 stimulates the reaction, probably by holding CMG to DNA for efficient use of its ssDNA gate (bottom right).
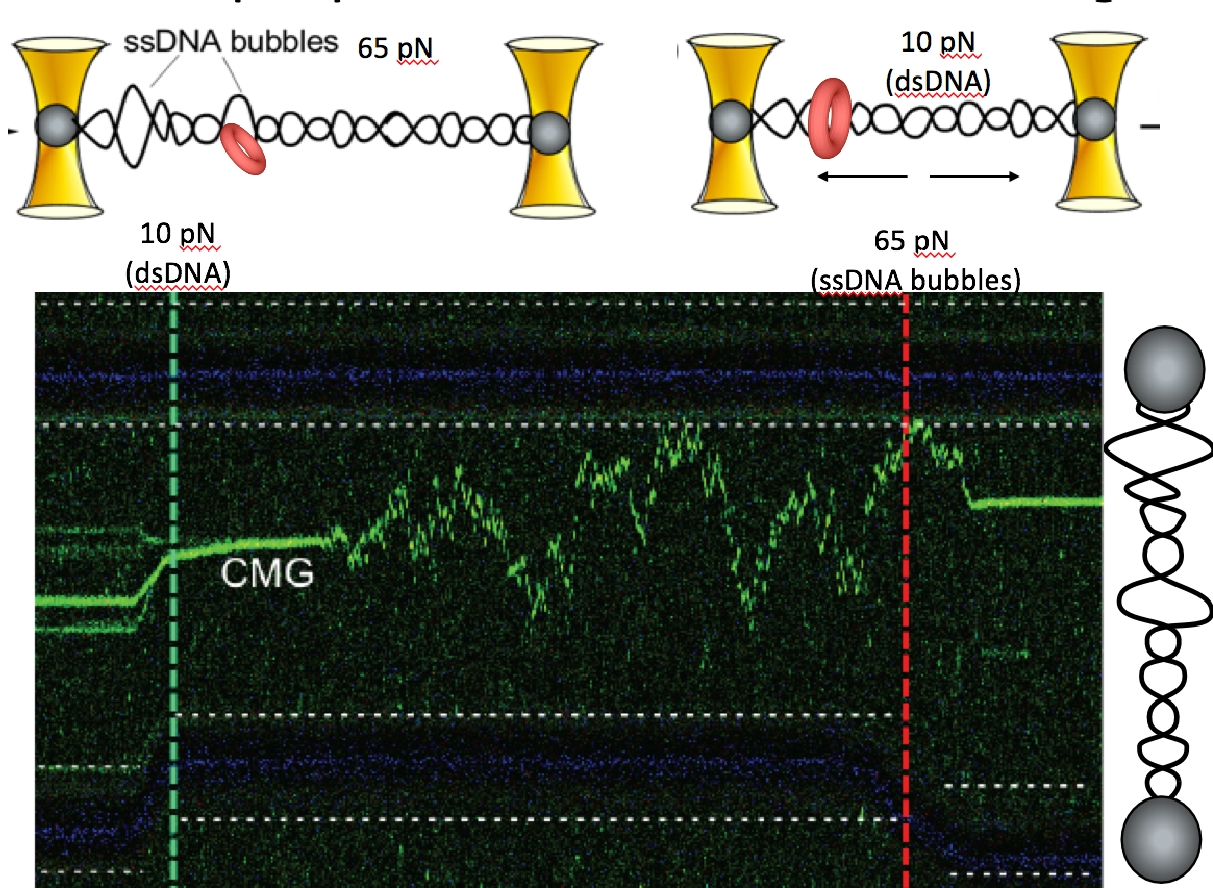
Figure 17. CMG can transition between dsDNA and ssDNA.At high (65 pN) force the CMG is at the forked juncition (left of green dashed line). But upon lowering the force to only 10 pN (right of the green dashed line), the CMG switches to a dsDNA diffusive mode. Upon elevating the force to 65 pN (right side of the red dashed line) new ssDNA bubbles form in the lambda DNA and the green CMG rapidly binds to the new fork.
For CMG to make this transition between dsDNA and ssDNA, the CMGs must expel one strand of the melted DNA, and thus require a ssDNA gate at one interface of the Mcm2-7 ring for the expulsion process. We have collaborated with Shixin Liu (Rockefeller University, NYC) to determine if CMG can switch between dsDNA and ssDNA using single molecule studies and fluorescent CMG, and whether Mcm10 is needed. This work is currently in BioRxiv, and under review for publication. Below is an illustration of the single-molecule method used, and some of the data from the BioRxiv submission (Figure 15). First, a ssDNA was stretched between two beads that were caught in separate optical traps (see Figure 15, top). Then CMG, labeled with a green fluorophore was added. The “green” CMG was able to assemble onto the ssDNA and track in one direction in an ATP dependent reaction (see Figure 15, bottom). The DNA tracking elements are located on the inside of all replicative ring shaped helicases, including CMG, indicating that the green fluorescent CMG encircles the ssDNA and explaining why they all track in one direction, dependent on ATP. Since the ssDNA is blocked on both ends by beads, the CMG is presumed to have a ssDNA gate to open and close around the ssDNA. The reaction was dependent on Mcm10 for efficient CMG loading onto the ssDNA. In fact, a ssDNA gate intrinsic to CMG was implied from the initial work on CMG from the Michael Botchan lab (UC Berkeley) who first purified and characterized CMG from the fruit fly. He found CMG could unwind an 32P-oligonucleotide from circular ssDNA, implying it has a ssDNA gate that enabled it to load on the circular ssDNA in order to melt the 32P oligo from the circular DNA. This type of assay is shown in Figure 16 for the budding yeast CMG +/- Mcm10. These experiments reveal the final steps of origin activation. The inward directed CMGs unwind DNA while encircling dsDNA, then a ssDNA gate enables CMG to evict one strand such that it encircles ssDNA. Both of these steps are greatly facilitated in the presence of Mcm10. We directly observed that CMG can transition from dsDNA to encircle ssDNA in the single molecule set-up in collaboration with Shixin Liu (Rockefeller University). When dsDNA is attached to beads, and a force of 65 pN is applied, ssDNA bubbles are produced upon which CMG can bind, as seen by the green CMG bound to the DNA at the left of the green horizontal line in the kymograph Figure 17. The the right of the green dashed line, the force is reduced to 10 pN, collapsiing the ssDNA forks to dsDNA, and this induces CMG to rapidly diffuse on the dsDNA. Upon re-introducing ssDNA bubbles by application of force (right of red dashed line), the diffusing CMG on dsDNA can transition onto ssDNA. We presume this occurs via a ssDNA gate for CMG to pass over the fork and encircle the leading strand. Indeed, upon adding replisome factors nucleotides, fork movement was observed to commence (not shown here).
Fork Reversal Repair
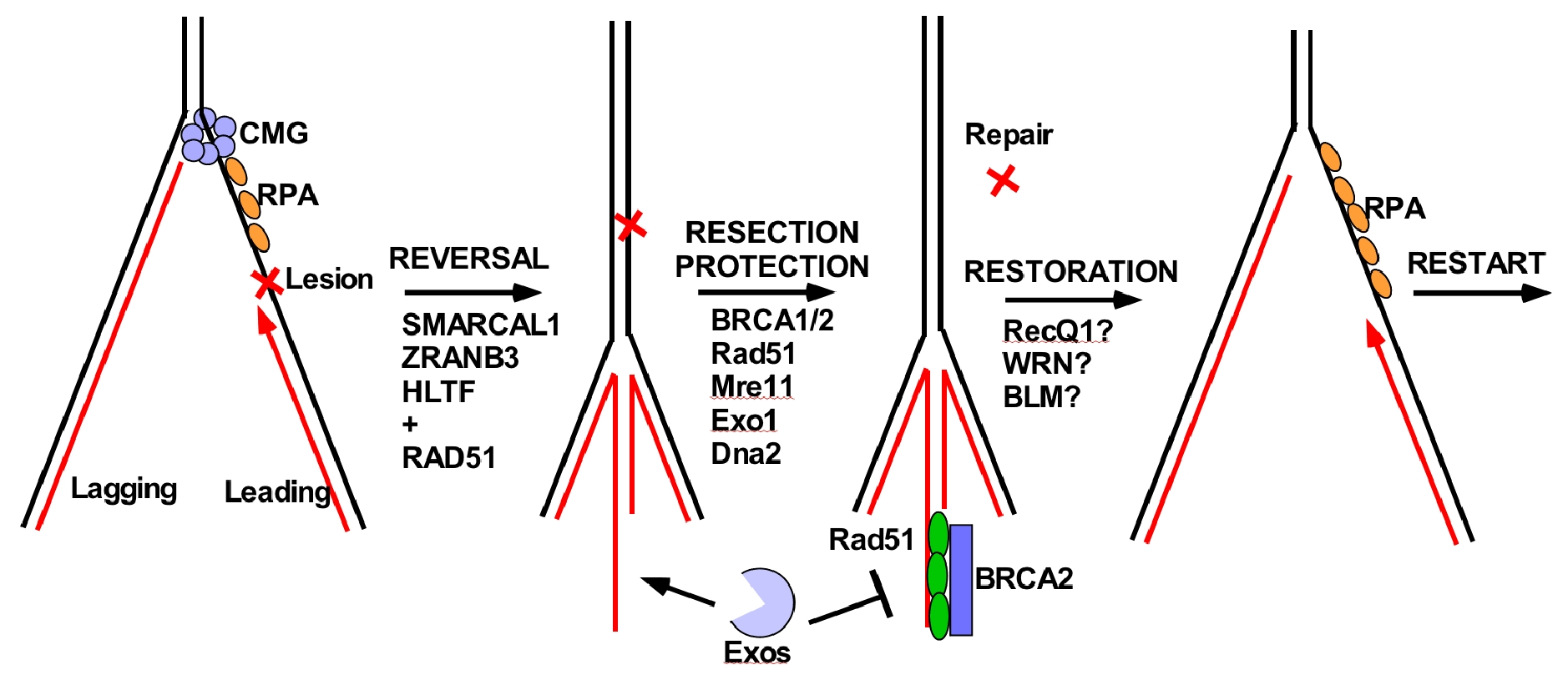
Figure 18. Fork reversal repair in eukaryotes.
Fork reversal occurs upon replisome stress, generated by lesions or encountering difficult to replicate sequences. In higher eukaryotes, fork reversal is known to be important to genome integrity. A diagramatic example of fork reversal is shown in Figure 18. Upon encountering a leading strand lesion, Pol epsilon will stall and the CMG helicase will depart from the Pol epsilon. DNA translocases then move the fork backward to form a 4th arm made from the two complementary nascent (newly synthesized) DNA daughter strands, which can be seen in the EM. In the example of Figure 18, this places the lesion into dsDNA, which can be acted upon by repair factors (not possible in the context of ssDNA). But, the 4th arm is percieved by the cell as a dsDNA break, and factors that fix double strand breaks come into play, referred to below as the “Resection/Protection” step. Specifically, nucleases like Mre11 and others, digest the 4th arm, but Rad51/BRCA1 bind the ssDNA produced by resection and protect against further excision. Lastly, the repaired fork undergoes restoration, to reanneal the nascent DNA strands to the parental strands and forming the 3 way replication fork junction.
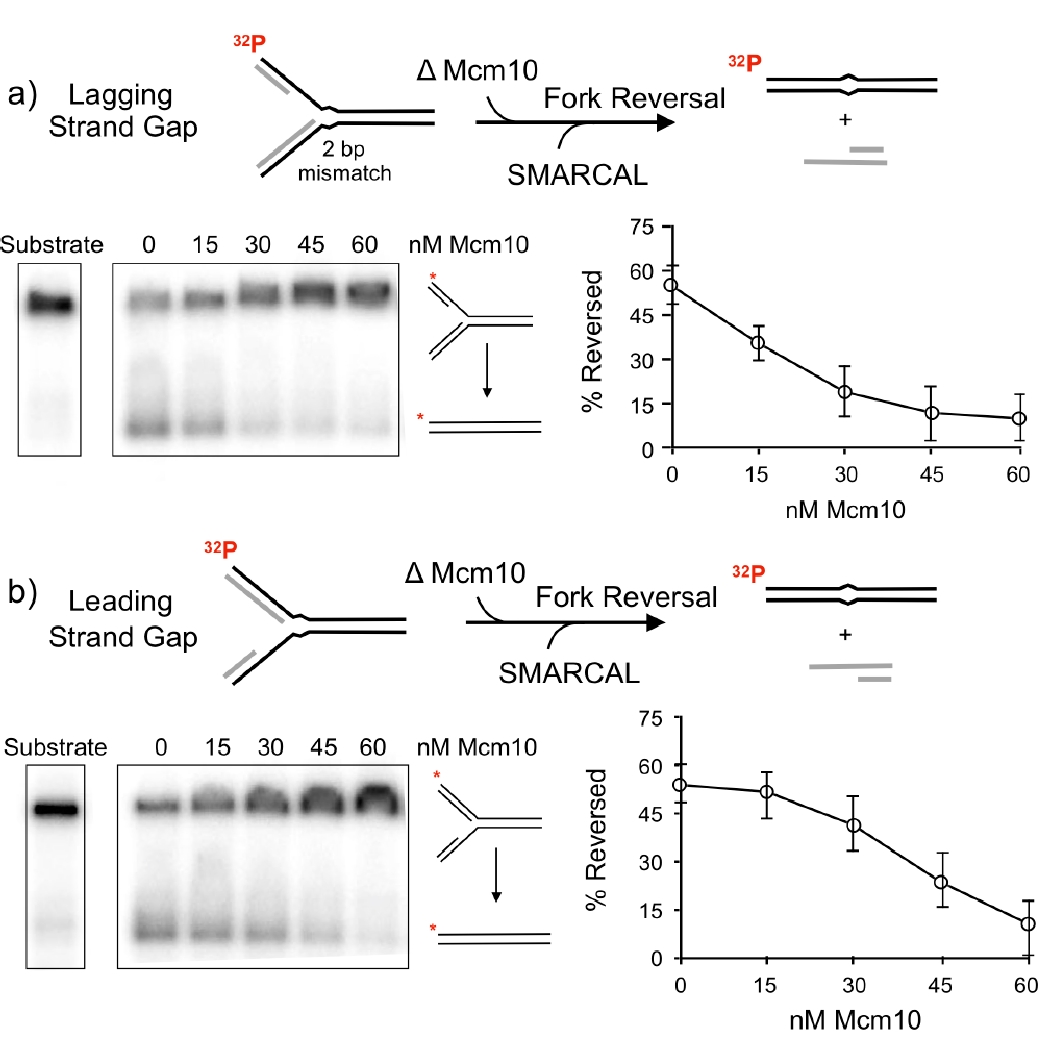
Figure 19. Mcm10 inhibits fork reversal by SMARCAL1. The native agarose gels show the substrate and product of fork reversal. The plots to the right show quantitation of Mcm10 inhibition of SMARCAL1 fork reversal.
Thus far, the role of the replisome or its fate during fork reversal have not been explored. We have taken a first step in defining what may distinguish an active fork from a fork that requres fork reversal repair. The fork reversal step is performed by either of three dsDNA translocases, SMARCAL1, ZRANB3 and HLTF. We have asked what prevents a functional replisome from becoming reversed? Now that we know CMG travels NTD first, we determined numerous contacts of the Mcm10 factor to CMG (i.e. Movie 5). The DNA binding domain of Mcm10 is located at the NTD of CMG near the central channel, indicating it is very close to the forked nexus, and thus could influence the ability of fork reversal enzymes to reverse actively moving forks. Using this piece of crucial intelligence, we determined whether Mcm10 could inhibit SMARCAL1 fork reversal activitiy. In fact, Mcm10 was highly efficient at inhibiting SMARCAL1 fork reversal. An experiment of the report that documented this is shown in Figure 19.
